SMARCB1缺陷型颅底软骨肉瘤?12p、IDH1/2检测确认睾丸生殖细胞肿瘤转移
时间:2025-02-13 12:15:13 热度:37.1℃ 作者:网络
体细胞型恶性肿瘤(STM)很少发生在原发性或转移性睾丸生殖细胞肿瘤(TGCT)中,并且与预后不良和生存率不佳有关。具有软骨肉瘤特征的STM极其罕见,并且之前没有报道过头部和颈部受累的病例。本文报道一名 39 岁的白人男性,因鼻塞和鼻出血就诊。影像学检查显示其鼻腔及颅底存在一个 6.9 cm的膨胀性肿瘤,并侵犯至眶内和颅内。组织病理学特征符合II-III级软骨肉瘤。免疫组织化学检查显示,恶性细胞对S100和上皮标志物分别呈强阳性和弥漫阳性,且SMARCB1表达缺失。未检测到IDH1/2突变。全身PET扫描发现左侧睾丸有一 7.0 cm肿块,诊断为伴合体滋养层细胞的精原细胞瘤,分期pT3NXM1b。还发现广泛的腹膜后、纵隔和锁骨上淋巴结肿大。左锁骨上淋巴结的组织病理学检查显示转移性精原瘤。FISH检测发现,大多数转移性淋巴结精原细胞瘤细胞携带 1-4 个i(12p)拷贝,而软骨肉瘤则以12p重复为特征。鉴于患者存在伴有弥漫性膈上淋巴结肿大的恶性TGCT、颅骨软骨肉瘤独特的免疫表型、12p扩增和IDH1/2突变缺失,均强烈支持将患者诊断为转移性TGCT引起的STM软骨肉瘤。患者对化疗没有反应,确诊三个月后去世。尽管极为罕见,但TGCT患者可能会转移至头部和颈部。与“典型”的头颈部软骨肉瘤相比,本文STM软骨肉瘤病例表现出不同的免疫表型和分子特征。对于具有典型组织形态但免疫组织化学或分子特征异常的病变,临床应保持高度警惕并明确其诊断。
背 景
软骨肉瘤家族是一类异质性恶性骨肿瘤,根据定义,其产生软骨(软骨样)基质,最常发生在骨盆、股骨、肱骨和肋骨。总体而言,软骨肉瘤约占所有肉瘤病例的 4% 和所有原发性骨恶性肿瘤的 11%。头颈部软骨肉瘤很少见,仅占所有人类软骨肉瘤的 1–12%,且好发于 40-60 岁的男性。头颈部的受累部位包括鼻窦、颌骨、喉和颅底,而疼痛、肿胀和鼻塞是最常见的临床表现。头颈部软骨肉瘤有多种组织病理学亚型,包括常规型、骨膜型、去分化型和透明细胞型,其中常规亚型主要见于颌面骨。在分子水平上,约 65% 的头颈部软骨肉瘤通常与异柠檬酸脱氢酶1/2基因(IDH1/2)的热点突变有关。
此外,据报道,具有软骨肉瘤分化的肿瘤与睾丸生殖细胞肿瘤(TGCT)中发生的体细胞恶性转化有关。体细胞型恶性肿瘤(STM)发生在 2.5%–8% 的TGCT中,仅发生于 15–68 岁(平均年龄:33 岁)的青春期后个体。这些最常见的原因是同时存在或先前存在的畸胎瘤,或与具有畸胎瘤成分的混合性GCT有关。一小部分STM似乎源自非畸胎瘤性TGCT,包括卵黄囊瘤和罕见的精原细胞瘤。STM可在睾丸原发性恶性肿瘤或淋巴结转移性肿瘤(通常是腹膜后和纵隔)或接受顺铂化疗后的其它器官中发展。转移性GCT中STM的发展严重影响预后和患者生存,而原发性肿瘤中STM的预后意义尚不清楚。判断转移性STM是源自TGCT还是涉及其它器官或组织的新发恶性肿瘤可能具有挑战性,尤其是当活检标本中未发现常规GCT时。
伴有明显软骨肉瘤特征的原发性或转移性TGCT相关STM的病例在英文文献中极为罕见,据本文作者所知,之前尚未有头颈部受累的病例。本文报告了颅底软骨肉瘤的临床病理学、免疫组织化学(IHC)和分子特征,这些特征有力地支持了转移性TGCT导致STM的诊断。
病 例
患者男,39 岁,白人,6 个月来鼻塞和充血逐渐恶化,偶尔出现鼻出血。既往因视力障碍而接受眼科检查,确诊双侧视盘水肿。MRI和CT扫描成像显示鼻腔中心有一个 6.9×5.3×5.0 cm的膨胀性异质肿块,延伸至前筛窦(图1A)。肿块表现出侵袭性临床特征,包括鼻中隔糜烂、所有鼻甲骨破坏并延伸至上颌窦,邻近骨质重塑、变薄和开裂(图1B)。可见肿瘤向眶内扩散,伴有相邻的纸质板重塑和中直肌畸形,以及向颅内扩散,伴有前颅窝底裂开,包括筛板和筛窦凹(图1A和B)。可见对视交叉后视神经有严重的占位效应。还可见蝶骨翼和翼突破坏,并累及圆孔、翼管和翼腭窝,以及鞍区和鼻咽部扩散。下方,肿瘤与硬腭相邻,无糜烂迹象。病变的临床和影像学特征提示可能为软骨肉瘤或脊索瘤,因此进行了活检。但患者左眼视力急剧下降,紧急接受手术,全切除鼻窦肿块,重建颅底并进行视神经减压术。术后患者视力很快恢复。显微镜检查显示骨浸润性软骨肿瘤,主要具有多叶结构和适当细胞密度(图1C和1D)。腔隙内含有圆形、椭圆形至多边形细胞,细胞质丰富,嗜酸性,光学透明或囊泡状,细胞边界清晰,周围为嗜碱性基质(图1C和1D)。肿瘤的其它区域细胞密度明显增高,大簇非典型、圆形至卵圆形和罕见细长细胞浸没在不同数量的嗜酸性或双嗜性软骨黏液样基质中(图1E和1F)。肿瘤细胞表现出明显的核多形性,单个细胞核从小的圆形到卵圆形,核轮廓光滑,染色质分布均匀(图1D),到较大的卵圆形或不规则形状,染色质开放,可辨别一个或多个嗜酸性大核仁(图1E和1F)。核内胞质常见假包含体和多核(图1F),有丝分裂象也常见,包括非典型有丝分裂(图1D和1F)。在病变细胞较多的区域,局部可见少数破骨细胞型多核巨细胞散在于恶性细胞中。未观察到骨样或未成熟骨形成的证据。上述组织病理学发现与II-III级常规型软骨肉瘤相符。
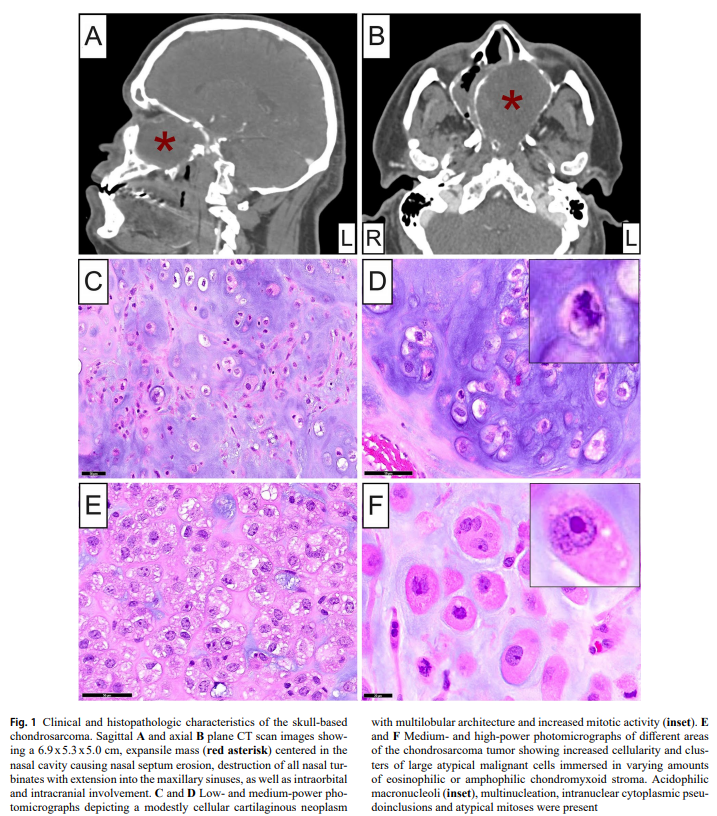
▲图1 颅底软骨肉瘤的临床和组织病理学特征
免疫组织化学检查显示,软骨肉瘤细胞对S100(核染色和细胞质染色均有;图2A)、波形蛋白、上皮膜抗原(EMA;图2B)呈强阳性,且呈弥漫性阳性,并与CAM5.2(图2C)和细胞角蛋白(CK)AE1/AE3局部免疫反应。有趣的是,SMARCB1(INI1)表达在恶性细胞中消失(图2D),但在背景内皮细胞和破骨细胞中保留(图2D)。病变细胞对brachyury、CK MNF116、高分子量CK、结蛋白、SOX10、OCT3/4和SALL4 均呈阴性。对从软骨肉瘤肿瘤中提取的基因组DNA进行焦磷酸测序,未在IDH1的第 132 个密码子或IDH2的第 172 个密码子处检测到突变。
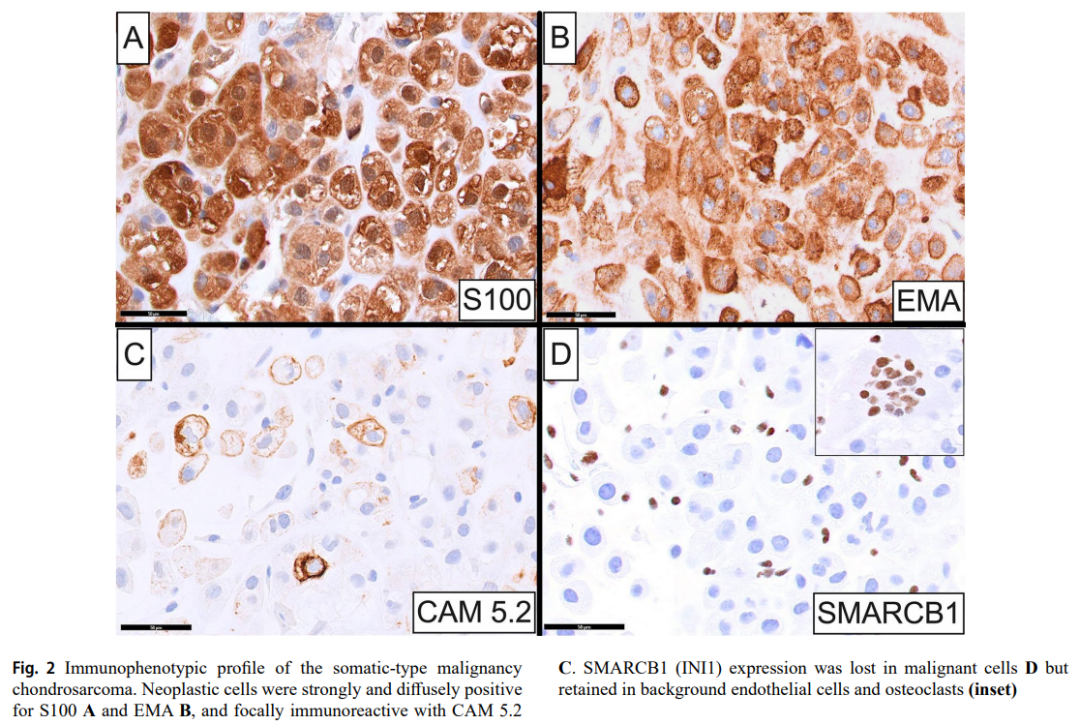
▲图2 体细胞型恶性软骨肉瘤的免疫表型特征
随后的全身PET扫描显示有轻度FDG摄取,左锁骨上淋巴结大面积代谢亢进,延伸至胸肌下和前纵隔区域,疑似转移性淋巴结受累(图3A)。此外,还发现明显的代谢亢进性股后、腹膜后和左主动脉周围淋巴结肿大,以及双侧髂链淋巴结肿大。肺实质、肝脏、脾脏和肠道未受累。值得注意的是,患者阴囊内还出现摄取明显(最大SUV = 12.1)、边界不清的异质性肿块(图3A,箭头)。阴囊肿块大小为 7.0×6.4×4.7 cm,与左侧睾丸相关。实验室检查结果显示乳酸脱氢酶和β-人绒毛膜促性腺激素(β-HCG)水平明显升高,但甲胎蛋白正常。进行了根治性腹股沟左侧睾丸切除术,并对左锁骨上(Virchow)淋巴结进行了穿刺活检。
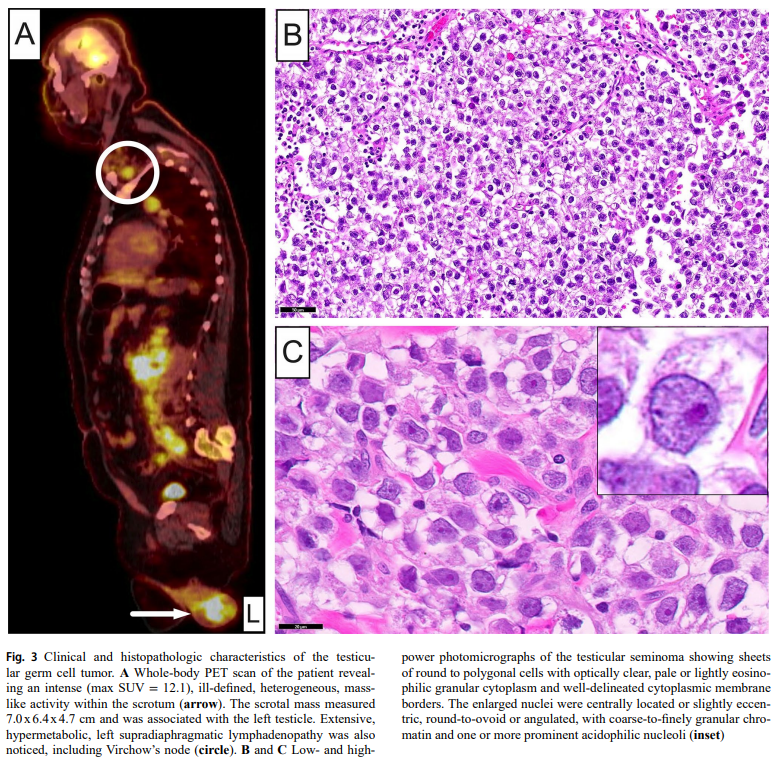
▲图3 睾丸生殖细胞肿瘤的临床和组织病理学特征
组织病理学上,睾丸病变由浸润性、均匀的恶性细胞群组成,这些细胞呈弥漫性片状排列,散布着大小不一的纤维隔膜(图3B)。在肿瘤的其他区域,可见结节性、嵌套性或假腺状结构。单个细胞呈圆形至多边形,具有光学透明、苍白或浅嗜酸性颗粒状细胞质和清晰的细胞质膜边界(图3B和3C)。细胞核增大、位于中央或略微偏心、圆形至卵圆形或有棱角,具有粗至细颗粒状染色质、一个或多个明显的嗜酸性核仁和不规则增厚的核膜(图3C)。存在散在的合体滋养细胞,以及大面积斑块状坏死、斑块状淋巴细胞浸润和淋巴血管侵袭。诊断为伴有合体滋养细胞的精原细胞瘤,分期为pT3NXM1b。在检查的任何组织病理学切片中均未发现畸胎瘤成分或其他组织学类型。对肿大的左锁骨上淋巴结进行组织病理学检查,发现正常淋巴结结构消失,取而代之的是精原细胞瘤细胞和散在的合体滋养层细胞(图4A)。IHC显示这些细胞对GCT标记物呈弥漫阳性,包括OCT3/4(图4B)、SALL4(图4C)、CD117(c-KIT;图4D)和D2-40,证实了转移性睾丸精原细胞瘤的诊断,而CAM 5.2(图4E)、CK AE1/AE3、磷脂酰肌醇蛋白聚糖3和β-HCG突出了合体滋养层细胞。SMARCB1核表达保留(图4F)。阴性IHC标记物组包括SOX10、melan-A、S100、desmin、CD30、高分子量CK、CK MNF116和EMA。
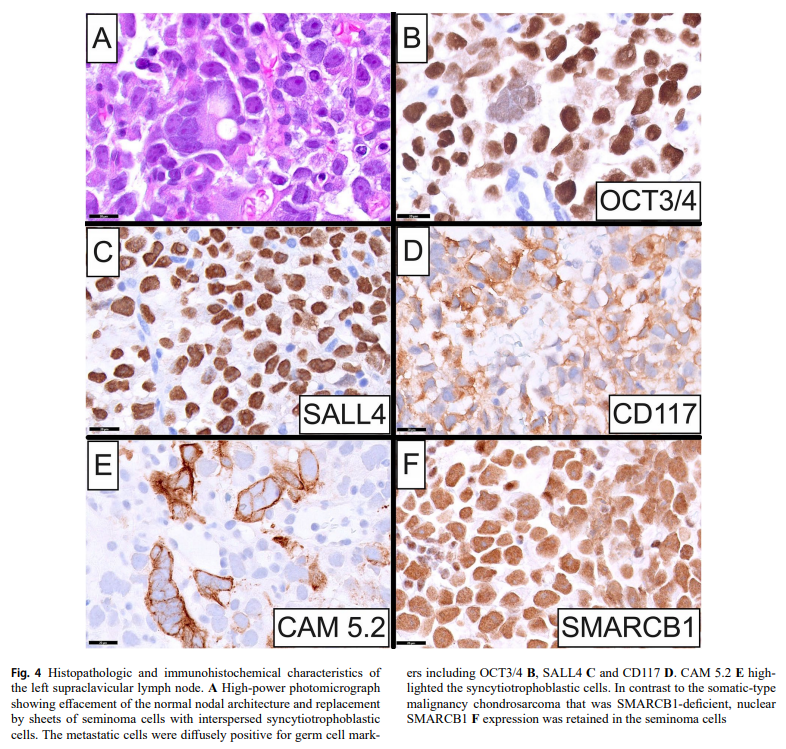
▲图4 左锁骨上淋巴结的组织病理学和免疫组织化学特征
鉴于发现恶性TGCT和患者弥漫性转移性淋巴结肿大,再加上颅底软骨肿瘤独特的免疫表型特征,即SMARCB1表达缺失,S100和EMA弥漫阳性,以及缺少IDH1/2突变,软骨肉瘤被解释为代表源自患者转移性TGCT的STM。进一步的FISH检测表明,大多数转移性淋巴结精原细胞瘤细胞中存在 1-4 个i(12p)拷贝,而软骨肉瘤细胞含有12p重复,从而证实了上述诊断。
患者接受了 4 个周期的BEP(博来霉素、依托泊苷和顺铂)联合化疗。治疗开始后三个月,即患者开始接受第 4 个周期的BEP治疗时,出现了剧烈咳嗽、呼吸困难加重和低氧饱和度。胸部CT检查显示右主肺动脉和同侧中下叶血管广泛肺栓塞。支气管镜检查和微生物培养显示凝固酶阴性葡萄球菌和草绿色链球菌,对此患者使用了广谱抗生素混合物。由于怀疑博来霉素引起毒性,患者还接受了增加剂量的泼尼松治疗。他出现了气胸,最终死于急性呼吸窘迫综合征和转移性TGCT肺栓塞引起的急性缺氧和高碳酸性呼吸衰竭。
讨 论
本文报告了一例诊断困难的软骨肉瘤病例的临床病理学、免疫表型和分子特征,该病例涉及一名先前未确诊的恶性TGCT和广泛淋巴结转移的男性的鼻腔和颅底。多项证据强烈表明,颅底软骨肿瘤是转移性TGCT的STM。正如研究人员在影像学和组织病理学上所发现的那样,患者表现为广泛的左侧转移性淋巴结疾病,涉及腹膜后、腿后、主动脉周围、纵隔甚至锁骨上淋巴结。总体而言,从膈下原发性恶性肿瘤转移到左锁骨上(Virchow)淋巴结并不常见,在TGCT病例中极为罕见。因此,鉴于膈上(包括锁骨上)淋巴结中存在转移性精原瘤,因此颅内和/或鼻腔鼻窦TGCT转移的可能性很高。
原发性或转移性TGCT内的STM的组织病理学诊断可能存在问题。根据WHO最新的分类,STM的诊断标准包括膨胀性过度生长,由纯粹的非典型间叶细胞或上皮细胞组成,占据至少一个低倍视野(4 倍物镜,直径 5 mm),并呈浸润性模式。在显微镜下,源自TGCT的STM表现出多种组织病理学类型,与TGCT形态学上不相似,包括肉瘤(约占所有病例的50%)、癌(20%)、胚胎型神经外胚层肿瘤(10%)、肾母细胞瘤,甚至髓系肿瘤和未分化肿瘤。值得注意的是,作为TGCT STM发生的癌症和肉瘤表现出不同的临床特征,前者在疾病过程中发展较晚(中位数:TGCT诊断后 108 个月),而后者通常出现较早(中位数:20 个月)。在肉瘤亚组中,横纹肌肉瘤占已报告的STM病例的 50% 以上。
在英文文献中,关于以传统软骨肉瘤为STM的TGCT的详细病例很少,这些病例要么以个案报告的形式发表,要么作为大规模临床病理学队列的一部分发表。在后者中,具有软骨肉瘤特征的STM仅占所有伴有STM的TGCT的 3.7%-5%。在之前报道的所有STM软骨肉瘤病例中(N = 5,年龄范围= 19-53 岁;平均年龄 = 32.2 岁),软骨肉瘤要么是在原发性睾丸肿瘤内发现的,要么在腹膜后淋巴结清扫术中发现,或者两者兼而有之。本病例是报道的第一例发生在睾丸外和结外解剖位置的STM软骨肉瘤。与大多数以前伴有STM的TGCT病例类似,软骨肉瘤发生于纯畸胎瘤、含有畸胎瘤成分的混合性GCT或卵黄囊肿瘤内。即使在大量取样后,STM也可能缺乏可识别的GCT成分,这增加了此类病例诊断的复杂性。在本文患者中,原发性睾丸肿瘤和淋巴结转移显示出具有合体滋养层细胞的精原细胞瘤特征,尽管在软骨肉瘤附近未发现精原细胞瘤成分。在极少数情况下,STM与“纯”精原细胞瘤有关。上述情况的一个合理解释是,在评估原发性睾丸肿瘤或畸胎瘤病灶时出现了取样误差,由于其体积小而无法进行组织学检测。
关于这种软骨肉瘤的另一个有趣发现是其独特的IHC特性,这进一步支持了其作为转移性TGCT的STM的病因。虽然肿瘤的整体组织形态学特征与软骨肉瘤的诊断完全一致,但病变细胞对S100和EMA表现出强烈而弥漫的免疫反应,以及CAM5.2和CK AE1/AE3的局部染色。鉴于传统软骨肉瘤不会明显表达上皮分化标志物,观察到的S100和EMA阳性使人们怀疑其为软骨样脊索瘤。然而,brachyury染色是一种对脊索肿瘤高度敏感和特异的标志物,均为阴性。GCT标记物OCT3/4和SALL4也呈阴性,而在转移性精原细胞瘤细胞中则呈强阳性。如之前报道的,STM通常表现出与原发性TGCT不同的免疫特征。
SMARCB1是SWI/SNF染色质重塑复合物的核心亚基,其基因组失活和核表达缺失是大量间充质肿瘤的特征,总体而言,这些肿瘤具有多样的组织病理学外观和生物学特性。SMARCB1缺陷型软组织肿瘤传统上由一群单一形态的未分化上皮样细胞组成,这些细胞具有“典型”横纹肌样细胞形态,以及间变性大细胞或小圆细胞特征。先前的多项研究表明,几乎所有恶性横纹肌样瘤和低分化脊索瘤、90% 的上皮样肉瘤以及一部分上皮样恶性周围神经鞘瘤、肌上皮癌和骨外黏液样软骨肉瘤均通过FISH或IHC检测显示SMARCB1失活和/或缺失。此外,SMARCB1胚系突变与家族性神经鞘瘤和脑膜瘤有关,在这些疾病中,获得性NF2突变与SMARCB1失活有协同作用。有趣的是,本文患者的STM软骨肉瘤通过IHC显示出均匀的SMARCB1阴性,但在非肿瘤背景细胞中保留了表达。据研究人员所知,之前仅报道过另一例SMARCB1缺陷型传统软骨肉瘤,患者为一名 13 岁男孩,肿瘤位于其下颌骨,有胸部恶性横纹肌样瘤病史,且存在潜在的胚系SMARCB1缺失。后者提出了一个问题,即本文患者是否也存在SMARCB1胚系变异,这将使他易罹患恶性肿瘤。然而,正如本文所显示的,转移性淋巴结精原细胞瘤细胞IHC染色显示,SMARCB1呈强阳性,这表明STM软骨肉瘤中SMARCB1的缺失代表获得性遗传或表观遗传现象,而不是胚系事件。
既往在 46.1%-71.4% 的颅底软骨肉瘤病例中发现了IDH1/2的遗传变异,近期则有 85.7% 的病例被发现。IDH1突变主要涉及4号外显子的第 132 个密码子,在约 40% 的病例中检测到R132C突变(CGT N TGT)。8.6% 的软骨肉瘤可见IDH2突变,其中大多数为R172S突变(AGG N AGT)。该软骨肉瘤缺少IDH1/2突变。尽管一小部分颅底软骨肉瘤可能IDH1/2变异呈阴性,但作为源自转移性TGCT的STM,这种肿瘤可能不是由常规软骨肉瘤的常见遗传机制驱动的。检测i(12p)或12p过表达是TGCT的标志性特征,已在大多数STM中得到证实。因此,通过FISH或定量PCR进行细胞遗传学研究以识别i(12p)可能对与常规GCT成分无关的STM病例(如当前的软骨肉瘤)具有诊断价值。值得注意的是,FISH分析证实了软骨肉瘤细胞中12p的异常重复,以及转移性精原瘤细胞中i(12p)的多个拷贝,从而验证了颅底软骨肉瘤是来自转移性精原瘤的STM。
识别与TGCT相关的STM至关重要,因为它们的存在会极大地影响预后和总体生存率。此外,正确识别STM组织病理学亚型可能具有治疗意义。尽管以顺铂为基础的化疗对常规TGCT非常有效,预后良好,但此类治疗方案对源自TGCT的STM通常无效。与此一致的是,本文患者对给予的博来霉素、依托泊苷和顺铂联合治疗没有反应。尽管采用了多模态化疗方案,STM患者的癌症特定生存率仍在 50%-60% 之间。
总之,本文报告了一个罕见的颅骨传统软骨肉瘤病例,该病例为一名 39 岁男性,表现为由转移性精原瘤引起的未接受化疗的STM。与“典型”传统头颈部软骨肉瘤病例不同,STM 软骨肉瘤显示SMARCB1表达丧失,上皮标志物(包括EMA、CAM5.2和CK AE1/AE3)呈局灶性至弥漫性免疫染色,以及12p重复,无IDH1/2突变。尽管极为罕见,但睾丸肿瘤患者可能会转移到头颈部。本病例报告还强调了对病变诊断高度怀疑的重要性,这些病变具有典型的组织形态学,但具有意外或不常见的IHC或分子特征。

▲摘自《睾丸生殖细胞肿瘤规范化标本取材及病理诊断专家共识》


▲摘自《卵巢恶性生殖细胞肿瘤临床诊治中国专家共识》
我司“生殖细胞肿瘤染色体拷贝数变异检测项目”,采用CNV-seq方法,覆盖了染色体12p获得或等臂变异,辅助区分睾丸青春期后型畸胎瘤和青春期前型畸胎瘤,鉴定睾丸实质原发的体细胞型癌(如腺癌和鳞状细胞癌等)和肉瘤是否与睾丸生殖细胞瘤相关。另外,还可以辅助卵巢生殖细胞肿瘤(无性细胞瘤和卵黄囊瘤)的临床诊断。
参考文献:
Argyris PP, Challa B, Satturwar S, VanKoevering KK, Wakely PE Jr. SMARCB1-Deficient Skull Base Chondrosarcoma with 12p Duplication Presenting as Somatic-Type Malignancy Arising from Metastatic Seminoma. Head Neck Pathol. 2024 Jan 18;18(1):1. doi: 10.1007/s12105-023-01610-5. PMID: 38236556; PMCID: PMC10796880.


