【醉翁之艺】围术期器官保护之五:组蛋白H3K18和Ezrin乳酰化促进脓毒症相关急性肾损伤的肾功能障碍
时间:2025-03-31 12:12:11 热度:37.1℃ 作者:网络
脓毒症相关急性肾损伤(SA-AKI),作为脓毒症患者极为常见的并发症,严重侵蚀肾脏功能。乳酸,作为糖酵解的关键产物以及无氧代谢的核心原料,凭借促进组蛋白乳酰化修饰,发挥着举足轻重的作用。组蛋白乳酰化,作为一种广泛存在的翻译后修饰形式,H3K18la 恰是其修饰位点家族中的重要成员之一。然而,组蛋白乳酰化作为表观遗传修饰在脓毒症相关急性肾损伤(SA-AKI)发展历程中的神秘面纱仍未被完全揭开。崔丽艳团队揭示了SA-AKI中H3K18和Ezrin-K263乳酸化水平升高,在乳酸与脓毒症肾损伤之间建立了联系,可能成为调节肾近曲小管上皮细胞表观遗传修饰和代谢的重要潜在治疗靶点,其结果发表于Advanced Science。

研究结果
SA-AKI肾组织和肾小管上皮细胞组蛋白乳酰化修饰迅速增加
为深入探究SA-AKI发生后,机体内乳酸以及乳酰化修饰水平所呈现出的变化情况,本研究作者运用动物实验与细胞实验相结合的方式,着重针对脓毒症后乳酸和组蛋白乳酰化修饰水平展开检测。研究结果显示,在经历SA-AKI后,乳酸水平与乳酰化修饰水平呈现同步升高态势。就组蛋白乳酰化修饰而言,仅组蛋白H3K18la在动物实验及细胞实验中均出现水平升高的情况。进一步针对不同浓度LPS刺激细胞后、在不同时间节点上乳酰化修饰所发生的改变进行探究发现:伴随LPS浓度逐步递增,总乳酰化水平以及H3K18la水平呈逐渐上调趋势。在LPS刺激后3小时,乳酰化水平开始升高;其中,HK-2细胞在LPS刺激12小时后乳酰化水平达到峰值,而TCMK-1细胞则于刺激后6小时达到峰值;至LPS刺激48小时时,乳酰化水平回落至基线水平。值得注意的是,在此过程中,乙酰化修饰水平并未出现明显的变化情况。具体来看,H3K18la水平在HK-2细胞中先是逐渐升高,不过在24小时开始下降,而TCMK-1细胞能够维持高水平的H3K18la直至48小时。
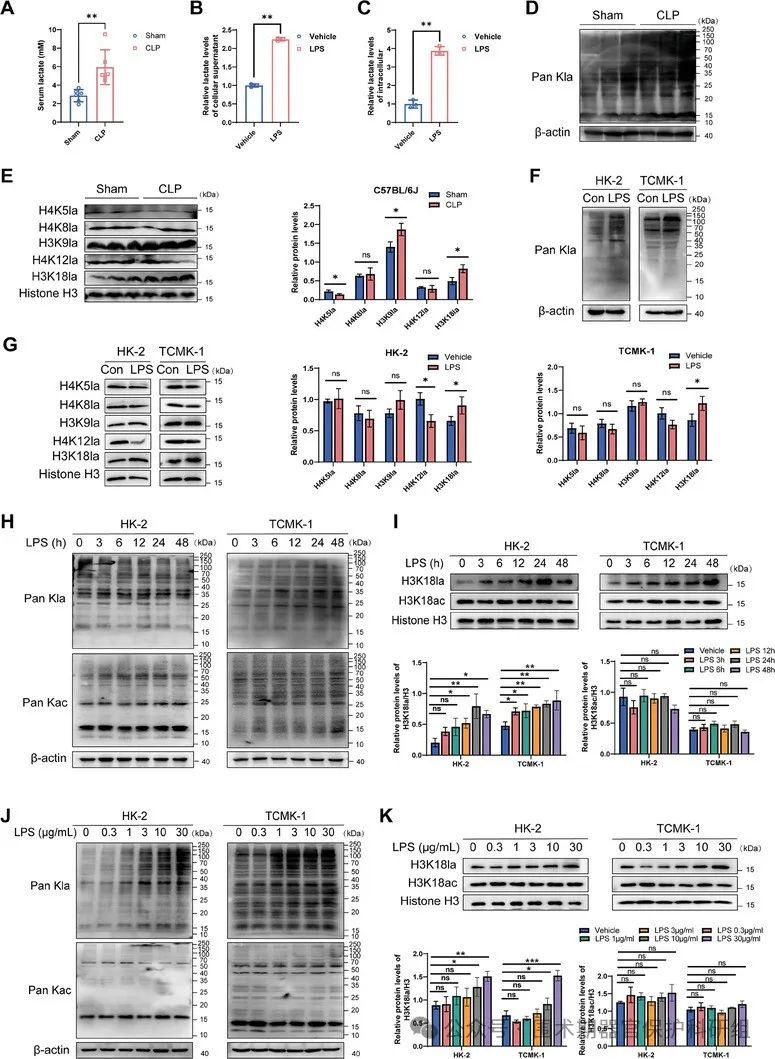
图1. LPS诱导SA-AKI小鼠和RTECs中组蛋白乳酰化增加
抑制组蛋白乳酰化可减轻LPS诱导的RTECs损伤
在利用葡萄糖转运蛋白GLUT1抑制剂(BAY876)对糖酵解过程实施干预抑制后,经检测发现,机体内乳酸水平以及组蛋白H3K18la水平均出现显著下降的情况,与此同时,氧化应激反应亦得到了有效的抑制。不仅如此,抑制糖酵解过程这一举措,还能够减少SA-AKI中细胞凋亡,并且可促使IL-6、IL-1β、TNF-α等炎症因子的浓度有所降低。
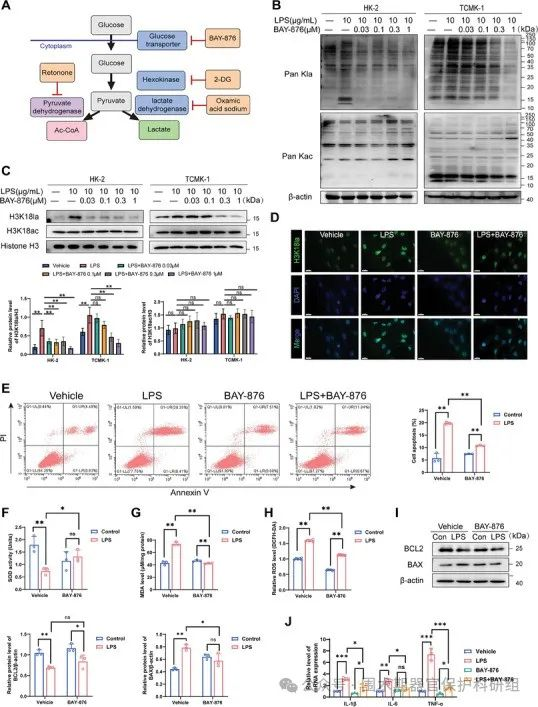
图2. 抑制GLUT1可降低组蛋白乳酰化水平,减轻LPS诱导的RTECs损伤
鉴定H3K18la调控的潜在下游靶点
组蛋白修饰在生物学过程中扮演着重要角色,其能够对靶基因的转录水平产生影响。鉴于此,为深入探究组蛋白H3K18la下游所涉及的调控机制,研究人员率先针对SA-AKI小鼠模型的全肾组织开展了ATAC-seq以及CUT&Tag分析。经过严格筛选,成功锁定了下游的若干关键基因,包括RhoA、Chac1、Akt1s1、Zfp36l1以及Scn1b。其中,RhoA 隶属于 GTPase 小家族,在脓毒症等多种炎症性疾病的发生发展过程中,对于细胞屏障功能的调节起着至关重要的作用。为对RhoA的转录是否与H3K18la呈现正相关这一假设进行验证,研究人员首先对比了RhoA区域CUT&Tag峰的信号情况。结果显示,相较于对照组而言,在CLP组的基因组相应位置,也即RhoA启动子处,H3K18la信号呈现出显著富集的状态。而值得注意的是,这种富集现象能够被葡萄糖转运蛋白GLUT1抑制剂(BAY-876)所抑制。不仅如此,经BAY-876处理后,还可观察到RhoA蛋白的表达水平出现了显著降低的情况。综合上述结果,由此确定RhoA为H3K18la的下游信号分子。
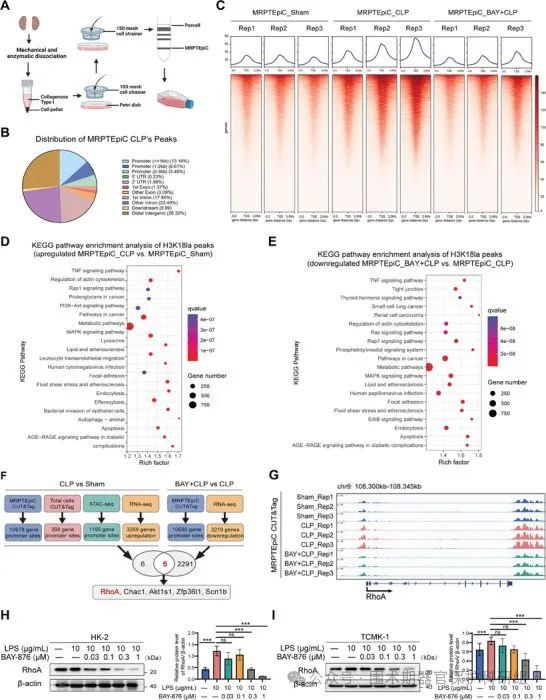
图3. 组蛋白乳酰化激活RhoA转录
LPS介导的RhoA-ROCK通路诱导Ezrin磷酸化和亚定位
为了研究RhoA/ROCK信号通路在LPS刺激的HK-2细胞中的潜在作用。 LPS诱导的RhoA和ROCK1蛋白水平在LPS刺激后3小时升高,6小时达到峰值,48小时后恢复到基线水平。ROCK参与了Ezrin的激活。Western blot分析显示,LPS处理的细胞中p-Ezrin蛋白表达水平显著升高,而敲除RhoA后p-Ezrin的上调被显著抑制。
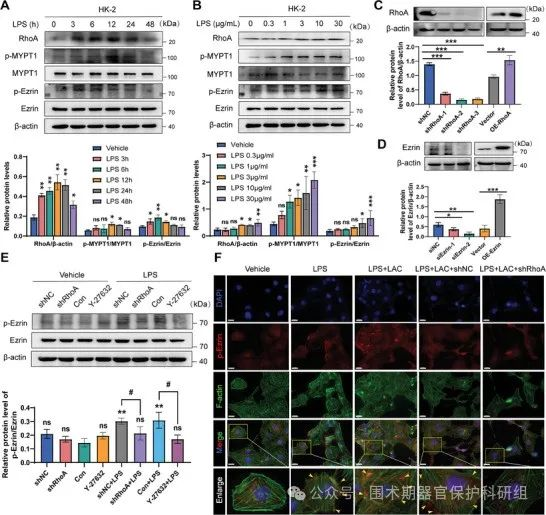
图4. RhoA/ROCK信号通路介导了LPS诱导的Ezrin磷酸化和易位
RhoA-ROCK1-Ezrin通路诱导NF-��B活化
为了研究RhoA/ROCK/Ezrin与炎症反应之间的关系。通过免疫共沉淀实验发现Ezrin与MyD88的互作关系。通过敲除和过表达Ezrin发现:Ezrin可以促进NF-��B的活化和细胞凋亡,同时也可以增加IL-6、IL-1β、TNF-α等炎性因子的水平。
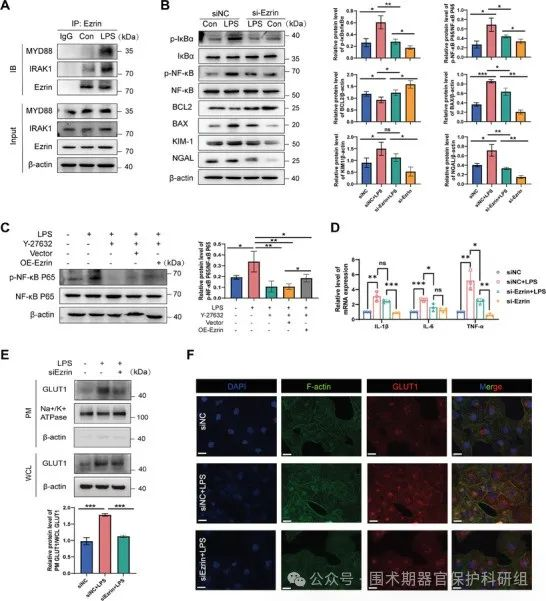
图5. Ezrin是RhoA的关键下游基因,调节RTEC损伤和GLUT1在质膜的易位
降低H3K18la具有抗炎作用,有助于SA-AKI后肾功能的改善
CLP后24小时,乳酰化水平显著上调,并一直保持在较高水平,直至72小时。H3K18la的乳酰化趋势与总乳酰化水平的变化一致。CLP后24小时免疫组化显示肾脏H3K18la水平显著升高,主要在肾近曲小管上皮细胞核中观察到乳酰化。BAY-876治疗(术前24小时,i.p.)抑制了肾脏整体乳酰化和H3K18la水平,降低RhoA、IKBα、NF-��B、IL-6和TNF-α的表达 ,同时可以改善SA-AKI肾功能。与BAY-876相比,乳酰盐(CLP后6小时,i.p.)处理后H3K18la水平显著升高。
实验结果充分表明,在SA-AKI这一病理过程中,组蛋白H3K18la水平呈现升高趋势时,会激活NF-κB通路,进而加剧肾脏损伤。与之相反,当组蛋白H3K18la水平有所降低时,则能够发挥抗炎功效,使得肾脏损伤程度得以减轻,并且有助于促进SA-AKI发生后肾功能的逐步改善,对肾脏功能的恢复起到积极的推动作用。
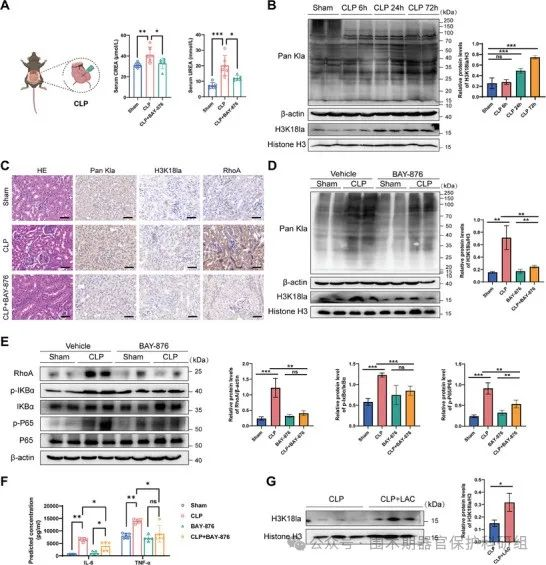
图6. 抑制GLUT1可降低H3K18la水平,改善SA-AKI后肾功能损害
乳酸促进Ezrin K263乳酰化,K263R突变逆转乳酸对fezrin介导的肾损伤的调节
液相色谱-质谱分析显示,Ezrin中乳酰化位点K3、K143、K162、K263、K450和K523可能存在显著差异。MOE分子对接预测了L-乳酸与Ezrin的结合亲和力,预测了氨基酸乳酰化的位点,K263R位点的突变显著降低Ezrin的乳酰化。Ezrin的三级结构表明K263位于Ezrin蛋白的FERM结构域,并且在多种哺乳动物物种中都是保守的。乳酸通过K263位点的FEzrin的乳酰化促进LPS诱导的肾损伤加重,K263可能是一个潜在的乳酰化修饰靶点。
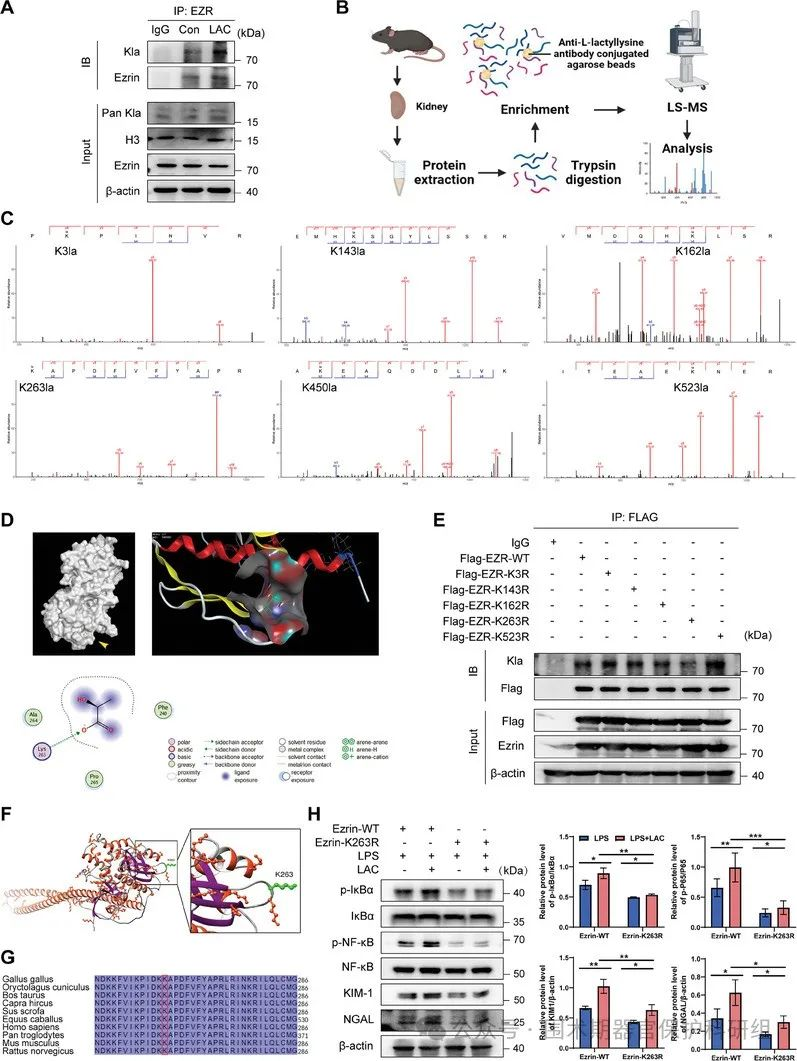
图7. 乳酸可促进Ezrin的K263乳酰化
研究小结
SA-AKI促进乳酸和H3K18la的增加,激活RhoA蛋白的表达,介导下游炎症和凋亡,导致肾损伤。相反,抑制糖酵解可减轻H3K18la的升高,为延缓肾功能不全提供了新的机制基础。糖酵解衍生的乳酸和H3K18la可能是改善SA-AKI肾功能的潜在靶点,未来可以通过降低NF��B激活来抑制RhoA/ROCK/Ezrin信号传导和Ezrin K263乳酰化导致的SA-AKI。
醉翁之艺 点评
在乳酰化修饰的研究领域中,围绕着乳酸诱导的底物蛋白乳酰化修饰主要包括两个研究方向:其一为组蛋白乳酰化修饰所介导的转录调控机制;其二是关于非组蛋白在发生乳酰化修饰后进而执行特定功能的探究。该项研究取得了重要发现,即乳酸所介导的组蛋白乳酰化修饰与非组蛋白乳酰化修饰均能对RohA-ROCK1-Ezrin信号通路产生调控作用,并且参与到脓毒症肾损伤的发生进程之中。具体而言,乳酸在这一过程中发挥了双重作用,一方面它能够通过诱导H3K18la调控RohA转录,进而激活RohA-ROCK1通路,上调Ezrin磷酸化,并最终激活NF-κB信号通路;另一方面,乳酸可促进非组蛋白Ezrin的乳酰化修饰,从而推动SA-AKI的发生与发展。本研究采用的针对组蛋白乳酰化修饰与非组蛋白乳酰化修饰对同一通路中上下游逐步进行验证的研究思路,极具借鉴价值。
此外,在该研究中,每一步实验均精心设置了浓度梯度以及药物刺激时长梯度,彰显出严谨的逻辑思维。研究借助动物实验来验证宏观层面的结果,通过细胞实验深入探索分子机制,二者相互结合有力地证明了乳酰化修饰在SA-AKI中的关键地位。并且,研究还通过糖酵解抑制剂治疗以及乳酸加重这两个方面,对SA-AKI过程中RohA-ROCK1-Ezrin信号通路的分子机制予以验证。未来,有望将糖酵解抑制剂作为一种潜在的治疗手段加以应用,同时也能够把H3K18la和Ezrin乳酰化修饰设定为靶点,以此来缓解SA-AKI患者终末期的病情进展。
总之,该研究着重聚焦于通过抑制糖酵解途径来减少乳酸生成,进而抑制乳酰化修饰以达成治疗的功效。然而,倘若能够通过加速乳酸的代谢来减轻SA-AKI 的病情发展,这无疑将会是一项极具前景与意义的研究方向,值得我们进一步深入探索与钻研。
参考文献
Qiao J, Tan Y, Liu H, et al. Histone H3K18 and Ezrin Lactylation Promote Renal Dysfunction in Sepsis-Associated Acute Kidney Injury. Adv Sci (Weinh). 2024;11(28):e2307216. doi:10.1002/advs.202307216.

