HR+早期乳腺癌血液ctDNA深度测序检出啥?ESR1/DDR/PIK3CA等耐药及转移相关基因突变
时间:2025-03-11 12:09:00 热度:37.1℃ 作者:网络
微创血浆游离DNA(cfDNA)分析能够为早期乳腺癌(EBC)提供基因组图谱,潜在识别与转移和治疗耐药相关的突变特征。在本研究中,我们使用定制的全面基因panel OncoIndx,对40例激素受体阳性(HR+)EBC患者的配对血浆和组织样本进行了测序。循环肿瘤DNA(ctDNA)基因组图谱显示,相较于肿瘤组织DNA(tDNA),其突变谱更为广泛,并且能够可靠地评估微卫星不稳定性(MSI)、肿瘤突变负荷(TMB)、同源重组缺陷(HRD)和杂合性缺失(LOH),这些均提示高度基因组不稳定性。值得注意的是,在ctDNA中早期检出雌激素受体α(ESR1)突变,突出了其识别有内分泌治疗(HR+乳腺癌的标准治疗方案)耐药风险的患者的潜力。DNA损伤反应(DDR)和增殖信号通路(磷脂酰肌醇-4,5-二磷酸3-激酶;PIK3CA)突变提示治疗耐药风险增加,为EBC患者的风险分层和个体化治疗策略提供机会。基于ctDNA的液体活检以微创的方式为EBC提供全面基因组分析,识别可干预的靶点和预测风险,从而更好地进行疾病管理。
研究背景
乳腺癌是全球范围内最常见的癌症之一,每年估计有230万新发病例。尽管癌症治疗取得了显著进展,改善了患者的预后,但仍有相当一部分早期乳腺癌(EBC)患者面临疾病进展、复发或治疗耐药的风险。激素受体阳性(HR+)的EBC,以激素受体的高表达为特征,通常预后较好。然而,尽管内分泌治疗具有临床应用价值,但仍有约30%的患者会产生耐药性,导致疾病复发或转移。了解EBC耐药的分子机制,对于改善临床预后至关重要,但目前仍面临诸多挑战。与转移性乳腺癌不同,下一代测序(NGS)技术在转移性乳腺癌研究中,为理解肿瘤生物学、亚型异质性和临床反应提供了有价值的见解,而EBC的基因组图谱却尚未得到充分的研究。与转移性疾病相比,早期疾病的NGS数据有限,这限制了我们对耐药和复发机制的深入了解,因此迫切需要可靠的生物标志物来指导个性化医疗。
基于组织的NGS基因组分析,虽然在评估突变状态方面具有重要价值,但只能提供肿瘤基因组的静态信息,无法反映肿瘤进化和疾病进展的动态变化。液体活检领域的重大进展,利用循环游离DNA(cfDNA)推动了包括乳腺癌在内的癌症治疗新方法的发展。这些技术能够无创检测肿瘤特异性的基因和表观遗传改变,为有效的疾病管理提供实时的临床信息。ctDNA是一种有前景的无创生物标志物,可用于检测微小残留病灶、监测治疗反应和预测复发。与传统的组织活检相比,ctDNA在确定肿瘤基因组图谱方面具有多项优势,包括能够全面捕捉肿瘤的异质性、跟踪突变谱随时间的动态变化,以及揭示治疗耐药的机制。最近的研究表明,ctDNA在确定EBC患者未来复发的高风险,以及理解转移性疾病的病理机制方面具有临床应用价值。
在转移性乳腺癌中,ctDNA已被证明是一种有价值的生物标志物,可用于跟踪肿瘤进化和检测耐药突变,从而根据肿瘤的基因图谱制定更有效的治疗策略。然而,在EBC中,由于临床认为疾病负荷较低,肿瘤DNA的脱落可能较少,ctDNA分析的作用尚不清楚。尽管ctDNA具有诸多优势,但目前EBC的研究主要集中在少数特定的突变或基因组改变上,很少考虑更广泛的突变谱在临床上的潜在价值。虽然使用小的靶向基因panel有助于检测热点典型突变,但往往对于低频出现的罕见亚克隆突变的缺乏灵敏度。此外,考虑到疾病处于早期阶段,大多数研究主要依赖基于组织的NGS来检测EBC中的致癌突变。
大量的临床前和临床研究表明,EBC在治疗过程中会发生广泛的分子和细胞进化。在治疗开始时,携带获得性突变的新克隆可能已经开始扩散。这些克隆群体可能使肿瘤细胞产生治疗耐药性,并提供生长优势。例如,ESR1基因的突变积累,会导致对抗雌激素或芳香化酶抑制剂维持治疗的耐药。由于组织活检存在取样偏差,以及受影响组织区域中肿瘤细胞的异质性,通过组织活检对新出现的突变进行分析存在一定的局限性。通过ctDNA分析及时检测这些新的或获得性的变异,能够为接受维持治疗的患者提供精确的治疗指导。
本研究旨在通过对一组EBC患者进行全面的ctDNA分析,填补这些研究空白。我们采用高灵敏度和高覆盖度的NGS方法,能够识别更广泛的突变谱,包括与疾病进展、复发和治疗耐药相关的已知和新发现的变异。值得注意的是,我们发现与以往研究相比,ESR1、NF1或NF2等耐药突变在人群中所占比例显著增加。我们进一步观察到,同时存在ESR1和PIK3CA或TP53突变的患者,无复发生存期明显缩短,并且出现早期转移进展。这些发现挑战了当前认为EBC中不会出现原发性耐药突变的观点,提示这些突变可能较为普遍,并且具有临床意义。更广泛的突变谱,有可能为风险分层提供依据,指导治疗决策,并识别出可能从替代治疗策略或更密切监测中获益的患者。例如,检测到ESR1突变(与HR+ EBC患者对内分泌治疗的耐药相关),可能促使医生选择替代疗法或联合疗法,以避免潜在的耐药问题。我们的研究强调了在EBC中进行全面ctDNA分析的重要性,证明了其在揭示具有临床可操作和耐药相关突变,以及指导治疗方法选择方面的潜力。
研究方法
根据制造商的说明,使用Sureselect XTSH2文库构建流程,从ctDNA和tDNA构建文库。简而言之,对ctDNA(50ng)和酶切片段化的tDNA(200ng输入)进行末端修复和poly A加尾。在目标富集之前,将包含8个碱基对的完全随机唯一分子标识符(UMI,其中包含索引)的接头连接到每个原始DNA片段上。使用靶向600个癌症相关外显子和选定内含子的杂交捕获定制OncoIndx® 600基因panel进行目标富集。在NextSeq 2000 NGS平台上以双端方式(150 X2)对捕获的目标进行测序,理论上ctDNA的测序深度为5500X,tDNA的测序深度为1500X。从ctDNA平均获得约1×10⁷ reads(范围:7×10⁶至12×10⁷),从tDNA平均获得约3×10⁶ reads(范围:3×10⁶至4×10⁶)。
使用iCare®生物信息学分析平台对原始序列reads进行分析。在比对(使用hg 38参考基因组)和一致性reads生成后,我们得到ctDNA的平均测序深度为1500X,tDNA的平均测序深度为500X。仅在检测所靶向的基因组区域中进行变异识别。突变的VAF定义为观察到的变异数除以总reads数。未通过预定义标准的变异被过滤掉。此外,同义变异和VAF低于0.1%的变异被排除在外。剩余的变异根据美国医学遗传学与基因组学学会(AMP/ACMG)进行分类,并进一步根据ClinVar和COSMIC数据库中的情况进行注释。只有被分类为致病性、可能致病性以及临床意义未明但潜在功能缺失(对于肿瘤抑制基因)或功能获得性(对于癌基因)变异,才被纳入后续分析。对于全基因组评分的MSI、TMB、HRD和LOH,分别采用以下阈值:MSI:MSI高≥20,MSI低<20;TMB:TMB高≥10,TMB低<10;HRD:HRD高≥42,HRD低<42;LOH:LOH高≥16,LOH低<16。
研究结果
40名激素受体阳性(HR+)EBC患者的55份血液和组织样本进行了全面基因组分析。在40名患者中,37.5%(n=15)和17.5%(n=7)的患者分别仅有ctDNA和tDNA样本,而45%(n=18)的患者有配对样本(同时提供血液和组织样本)。所有进行基因组分析的原发性肿瘤均未接受过化疗或在标准治疗的早期阶段。
从ctDNA获取的全基因组不稳定性特征
ctDNA的水平在不同样本之间存在显著差异。假设1 ng的ctDNA相当于300个单倍体基因组,我们在患者中观察到每毫升血浆中含有450到5930个单倍体基因组当量(GE)。ctDNA的水平可能因组织来源、肿瘤负荷和肿瘤的脱落特性而有所不同。在2000 GE的阈值下,ctDNA将样本分为两个不同的群体。ctDNA的平均浓度约为2040 GE(6.8 ng)/毫升血浆。这些样本中的肿瘤分数(肿瘤DNA占基因组当量的百分比)从17%到46.7%不等,平均值为34.3%。ctDNA GE与突变数量无关。然而,肿瘤分数(TF)较高(TF阈值≥25%)的样本具有更多的突变。当仅考虑高TF(≥25%)的样本时,TF与血液肿瘤突变负荷(bTMB)之间观察到弱相关性。
图1A展示了33名患者的ctDNA样本的MSI、HRD和LOH评分的分布情况。所有患者的MSI均为低(81.25%)或稳定(18.75%)。图1B展示了人群中TMB评分的分布情况。10名患者(25%)的bTMB较高,12名患者(30%)的bTMB评分较低。其余患者的bTMB评分接近阈值(10个突变/MB)。高bTMB人群(平均18.35)和低bTMB人群(平均5.6)的bTMB评分差异显著(P=0.0001,图1C)。此外,高bTMB人群比低bTMB人群具有更多的致病性或可能致病性突变(P=0.001)。
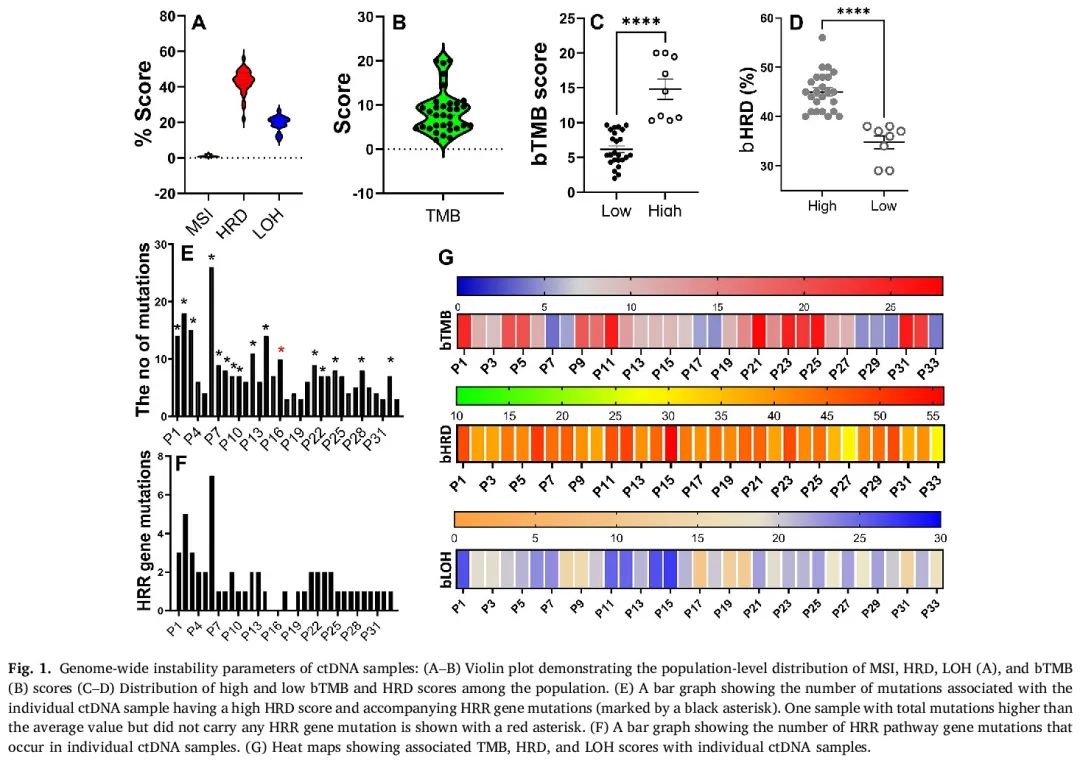
图1
我们观察到75%(25/33)的ctDNA样本具有高HRD评分(图1D)。所有高HRD评分的样本至少存在一个同源重组修复(HRR)通路突变,提示HRR通路活性异常(图1E - F,F检验,P=0.01,优势比:27)。在所有HRR通路突变中,BRCA1和BRCA2突变占比达75%。此外,BRCA1或BRCA2突变与bHRD评分的关联作为一个独立因素在BRCA阳性样本(13名患者)中显示出良好的相关性(F检验,P = 0.017,优势比:16.6,CI = 2.9至75.8)。我们进一步评估了高综合bHRD评分是否与致病性(P)/可能致病性(LP)突变的总数、bTMB,以及其他基因组不稳定性参数(如LOH、大片段迁移(LST)和端粒等位基因不平衡(TAI))相关。多变量分析显示,HRD与其他参数之间存在显著相关性(P=0.001;置信区间:0.017至1.3),但与bTMB评分无关。尽管bTMB与HRD之间的关联未达到统计学显著性(P=0.061),但两者之间存在正相关趋势。图1F展示了个体患者中检测到的ctDNA突变数量的分布情况。除了一个样本外,其他所有突变数量超过7个的ctDNA样本都与高HRD评分和HRR通路的突变相关(图1E)。图1G热图描述了个体ctDNA样本的bTMB、HRD和LOH评分情况。
ctDNA突变图谱揭示独特的突变图谱
在研究人群中,我们在ctDNA样本中共检测到250个P和LP突变,平均每名患者有8个突变。单核苷酸变异(SNV)占比较高,占总体突变的46.5%。由于终止密码子获得导致的无义变异(32.5%)在75%的患者中存在,而剪接变异(9.5%)、拷贝数变异(CNV,7.7%)和移码变异(3.8%)分别在25%、12%和25%的患者中检测到(图2A)。图2B热图展示了基于突变类型的患者间变异分布情况。约81%(26/32)的ctDNA样本同时存在SNV和无义变异,而SNV、截断变异和剪接变异仅在31.25%(10/32)的样本中单独出现。总体而言,SNV和无义突变的比例超过移码和CNV变异。图2C热图展示了考虑各个突变的变异等位基因频率(vaf)值时,检测到的突变总数的患者间分布情况。根据ClinVar和ACMG数据库,约22%(7/32)的患者ctDNA中存在超过10个P或LP突变。59%(19/32)的患者有5到10个具有临床意义的突变,其余19%(6/32)的患者ctDNA中的突变少于5个。
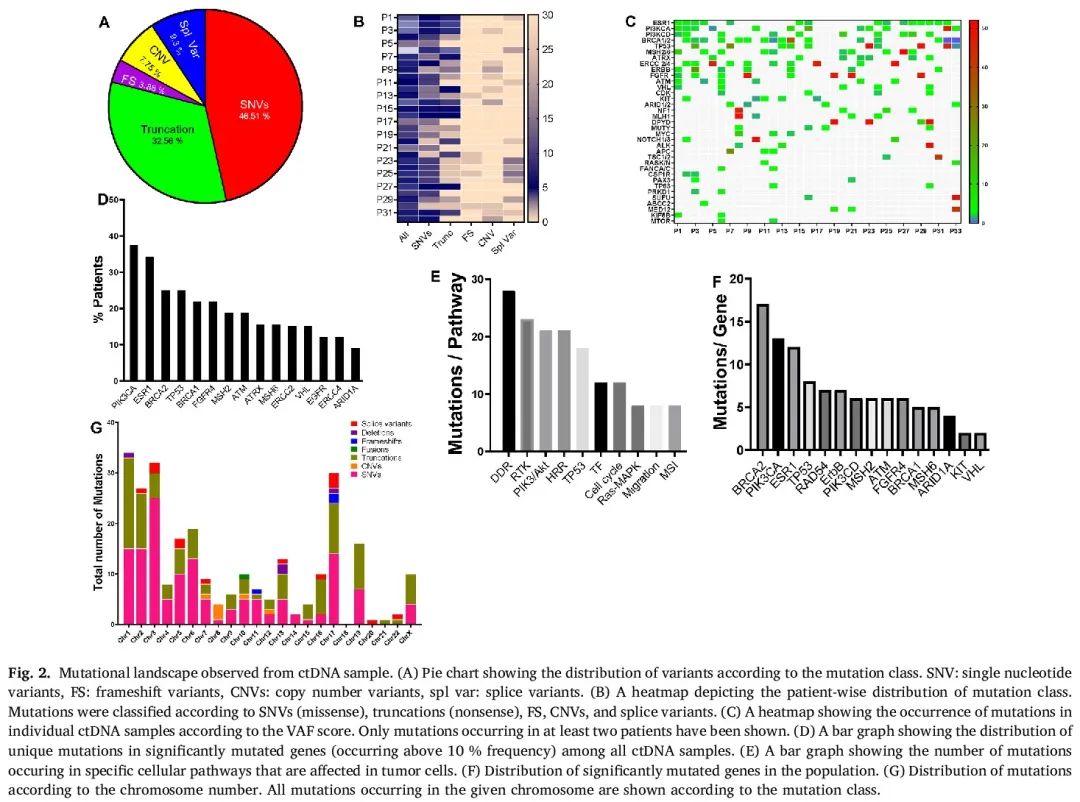
图2
图2D展示了人群中单个基因突变的分布情况。PIK3CA是最常检测到的变异(38%),其次是ESR1,在34.5%的患者中出现。循环肿瘤蛋白P53(TP53)变异在28.1%(9/33)的样本中检测到,随后是乳腺癌1型(BRCA1)和乳腺癌2型(BRCA2)变异,发生频率分别为21.8%和25%。除了ESR1外,这些发现与先前的报道相符。共济失调毛细血管扩张突变(ATM)、X连锁α地中海贫血/智力低下综合征(ATRX)、MutS同源物2(MSH2)、切除修复交叉互补(ERCC2)、Von Hippel - Lindau(VHL)和富含AT交互结构域1A(ARID1A)变异的发生频率均为10%(6/33)。我们还在32%(5/33)的患者中检测到二氢嘧啶脱氢酶(DPYD)的潜在致病性胚系变异,提示其可能更容易出现化疗毒性增强。在BRCA(BRCA1和BRCA2)阳性患者中,4名患者(4/15)携带胚系BRCA突变(BRCAness),并且这些突变与其他致病性错义变异同时出现。
我们进一步分析了人群中最常突变基因的共存情况。ESR1和PIK3(PIK3CA和PIK3CD)突变在28.1%的患者中同时出现(P=0.5,置信区间:0.56至0.57)。综合来看,在25%的患者中,循环ESR1变异与BRCA(BRCA1和BRCA2)变异同时出现。在最常突变的基因中,BRCA2的突变数量和多样性最高(图2E),其中截断、移码或缺失突变占主导(70.5%),这些可能导致功能缺失。PIK3CA突变中,单核苷酸多态性(SNP)的频率较高(92.3%),包括螺旋结构域和激酶结构域中经典的热点E542(外显子9)和H1047(外显子20)。ESR1突变主要是SNV(83.3%),包括配体结合域中N532、V534和P535(均位于外显子10)功能获得性错义突变,这些突变可能导致ESR1的配体非依赖性激活。有趣的是,72%的循环ESR1变异阳性样本同时具有高HRD评分。TP53突变大多是SNV和截断变异的混合,可能导致功能缺失。肿瘤抑制基因,如RAD54样(RAD54L)、ATM和BRCA1,大多发生无义突变,导致功能缺失。
基于ctDNA突变谱,我们根据受影响的细胞通路对变异进行分类(图2E)。观察到的大多数突变来自DNA结合蛋白基因(40%),其中18%与DDR相关,11%来自P53通路。其次是调节增殖信号的基因突变,包括受体酪氨酸激酶(RTK)和PIK3通路(占观察到的总突变的27%)。HRR通路相关变异的发生频率为13%,其中包括与DDR通路重叠的变异。有趣的是,7.5%的变异来自细胞迁移机制,这可能提示这些患者的亚克隆肿瘤细胞获得了侵袭性行为。在根据突变类型对染色体进行分析时,发现1、2、3和17号染色体的突变富集和多样性显著(图2G)。这些染色体共包含40%的变异,其中错义和无义突变占比最高。总体而言,以及在染色体层面,SNV(错义突变 + 无义)的占比显著高于其他突变(P=0.0186)。当考虑P或LP改变时,SNV分布在所有染色体上,而CNV和移码事件仅限于6条染色体。
tDNA的全基因组不稳定性特征
所有提供tDNA(FFPE来源的肿瘤DNA)样本的患者(n=22),根据组织病理学观察,肿瘤组织含量至少占20%。患者之间的肿瘤组织含量差异显著,肿瘤分数(肿瘤DNA含量百分比,tDNA)在30%到80%之间,平均值为54.8%。图3A展示了人群中MSI、HRD和LOH评分的分布情况。与ctDNA观察到的情况类似,tDNA样本间的MSI评分差异不显著(32% MSI低,68% MSI稳定)。HRD和LOH评分范围分别为22%-58%(平均40.2)和10-23%(平均16%)。与ctDNA相比,所有tDNA样本的TMB(平均=12.9)分布更窄,35.5%的样本具有高TMB评分(平均=19,图3B),提示与ctDNA具有良好的一致性。高TMB和HRD人群的平均TMB和HRD评分,与低TMB和HRD人群相比,差异显著(图3C,P=0.0001;图3D,P=0.0005,U检验)。图3E展示了TMB、HRD和LOH评分的患者间分布情况。tDNA的HRD评分分布更广,范围在20%到60%之间。低HRD评分(平均32%)见于43.5%的人群,而高HRD评分(平均46%)在56.5%的样本中观察到(P=0.0001,U检验)。在30%的样本中观察到HRR基因突变,且与高HRD评分相关。令人惊讶的是,50%的没有HRR突变的样本存在TP53突变。其中,75%的样本HRD较低,这可能提示相关的TP53功能缺失变异可能导致DDR通路失调加剧。这一观察结果与ctDNA队列形成对比,ctDNA队列中大多数样本存在HRR通路基因突变。所有tDNA样本的LOH分布较窄,低LOH和高LOH评分人群的一部分与各自的HRD评分相关性良好(R=0.7),提示它们之间良好的关联。在个体患者层面进行比较时,31.8%的患者至少有5个突变,50%的患者至少有3个突变。仅有2个样本观察到至少1个突变(图3F)。在比较人群中单个基因变异的分布时(图3G),PIK3CA突变最为常见,见于47%的人群,其次是TP53(31%)、ESR1(20%)和ERCC2(15%)。
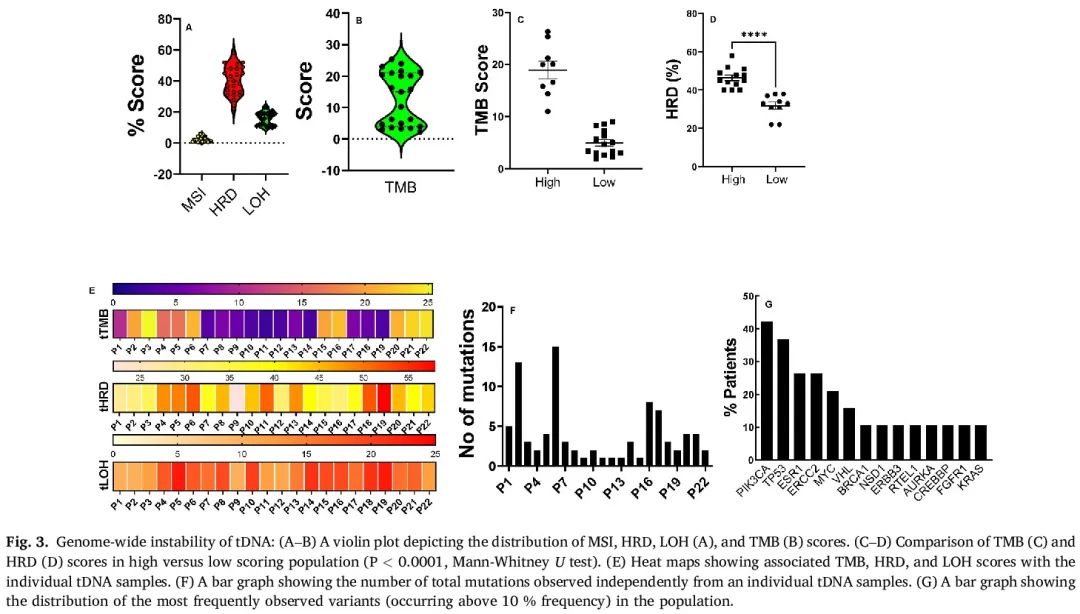
图3
tDNA突变图谱
我们共观察到113个突变,包括P、LP和临床意义未明的变异。与ctDNA类似,SNV主导了突变图谱,占所有检测到的变异的52.58%。其次是CNV(22.68%)、截断变异(14.43%)、移码变异(6.19%)和剪接变异(4.12%)(图4A)。与ctDNA样本相比,具有临床意义的变异的vaf的患者间分布差异显著(图4B)。tDNA样本中观察到高vaf评分,或可归因于从FFPE标本的肿瘤富集区域采集肿瘤细胞。根据突变类型进行分层时,我们观察到与ctDNA不同的变异模式(图4C和D)。令人惊讶的是,tDNA中检测到的所有CNV均为扩增事件,这与ctDNA样本不同。此外,截断事件在tDNA中的占比显著低于ctDNA(56%)。91%的样本中检测到SNV,而CNV和移码事件分别在50%和18.2%的样本中观察到。截断变异的总体占比低于CNV,但它们分布均匀,见于55.54%的样本。22.72%的样本中检测到剪接变异,且在单个样本中发生频率相同(所有剪接变异阳性样本均为1个变异)。50%的样本中同时存在SNV(错义变异)和无义变异,而40%的样本中SNV和CNV相互排斥。31.8%的tDNA样本中同时观察到SNV、CNV和错义变异。根据Clinvar和ACMG数据库,基于临床意义分组时,95.45%的样本至少携带1个P突变,54.54%的样本至少有3个P或LP突变。近59.1%的样本至少有1个临床意义未明的突变。与ctDNA相比,tDNA中变异的染色体分布模式不同。根据突变类型进行分类时,5、6、19和20号染色体的变异多样性较高。与ctDNA不同,变异在3、8、12和17号染色体上富集(图4E)。在根据突变类型分组后,ctDNA和tDNA变异在染色体分布上没有明显趋势。
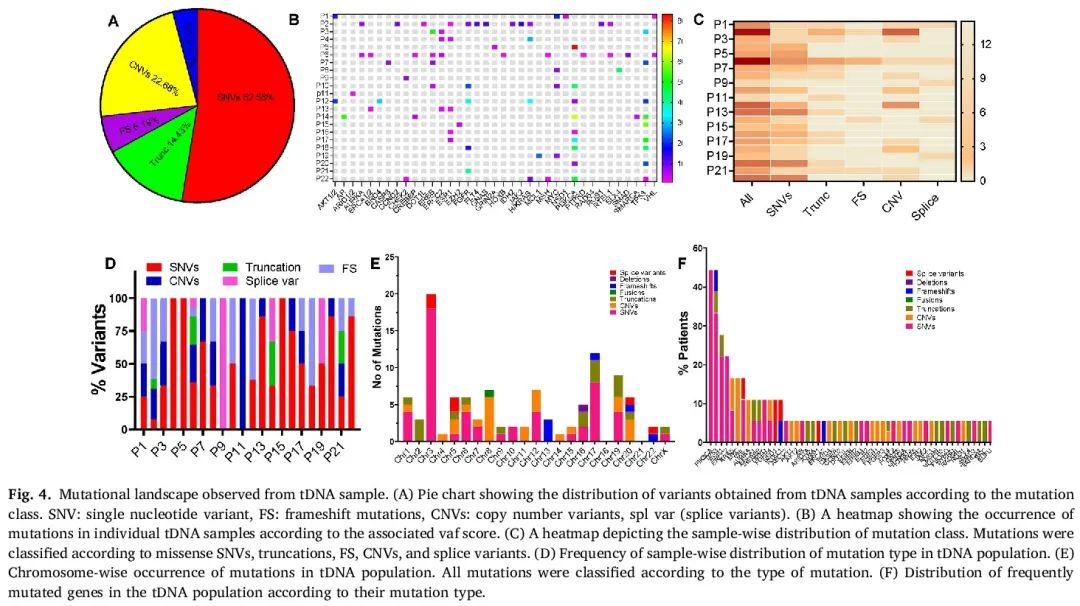
图4
分析单个突变特征时,PIK3CA是最常突变的基因,在45.45%(10/22)的人群中富集(图4F)。TP53是第二常见的突变基因,在人群中的频率为36.36%。出乎意料的是,仅在22.7%的人群(5/22)中检测到BRCA突变,其中为BRCA1突变。tDNA中ESR1变异频率为22.7%(5/22),而在ctDNA中频率为45%。其次是ERCC2和ERCC4变异,频率为13.6%,随后是MMR和RTK基因(合并)突变,在13%和36%的tDNA样本中存在。仅在1个tDNA样本中检测到DPYD胚系变异,该样本有配对的ctDNA,也存在相同的循环DPYD变异。在所有PIK3CA阳性样本中,80%为经典热点突变(分别为外显子10和21的E542K/545K和H1047R点突变)。除了2个样本外,当PIK3CA变异存在时,基于最大vaf评分,它是宿主肿瘤的主要驱动因素。
共突变分析显示,与ctDNA谱相比,呈现出不同的趋势。PIK3CA和TP53变异仅在9%的患者(2/22)中同时出现,提示在大多数样本中,两者具有较高的相互排斥可能性,或者在共存人群中存在亚克隆事件(P=0.2,优势比=0.25)。PIK3CA和ESR1变异在18%的样本中同时出现,这可能提示宿主肿瘤主要由失调的细胞增殖信号通路驱动(P=0.1,优势比=6)。PIK3CA和ESR1共突变与侵袭性生长和治疗耐药导致的治疗反应不佳有关。18%的患者同时观察到TP53和ESR1突变,且与高LOH评分相关。同时携带TP53和ESR1变异的肿瘤,往往基因组不稳定性较高,积累额外的突变,促进疾病进展,导致更差的临床结局。
从功能层面进行分类时,大多数突变表明增殖或促有丝分裂信号失调是主要的致瘤事件。约60%的样本存在激活和/或影响增殖信号通路(如PIK3、MAPK、RTK和生长因子受体(GFR)信号级联)调控的突变。数量最多的单个突变(20%)集中在RTK/GFR信号通路,其次是DDR(18%)、PIK3(11.8%)、HRR(10%)和TP53(5.8%)通路,其余的分布在代谢活动、染色质重塑和DNA结合蛋白中。令人惊讶的是,与ctDNA样本相比,tDNA样本中细胞周期调控机制相关突变较少,并且影响MSI和迁移通路的总突变数量在tDNA群体中几乎可以忽略不计。
配对样本中ctDNA展现更广泛和丰富的全基因组特征及突变图谱
在所有ctDNA和tDNA样本中,45%(18/40)为配对样本。在比较全基因组不稳定性参数(TMB、HRD和LOH)时,我们发现配对的ctDNA和tDNA之间的一致性为70%。50%的配对样本TMB评分较高,27.7%的配对样本TMB评分较低,一致性为83.33%。同样,配对样本之间HRD评分的一致性为66.66%,其中77.7%伴有高HRD评分。在比较LOH时,72.2%的配对样本具有一致性,其中83%的评分较高。在比较个体基因组瘢痕特征(LST + TAI综合评分)时,我们也观察到了较高的一致性。综合所有特征,配对样本之间的一致性达到70%或更高。对配对样本进行的多变量分析显示,HRD与其他基因组不稳定性参数显著相关,提示HRD评分是临床和治疗考虑的重要生物学指标。
随后,我们在突变水平上分析了配对样本的一致性。图5A展示了根据突变类型,ctDNA合并tDNA变异的分布情况。在配对样本中,SNV主导突变图谱(52.94%),其次是CNV(26.83%,包括扩增和缺失事件)。截断变异占比第三,随后是剪接变异(26.83%)和移码变异(2.94%)。图5B展示了配对样本中ctDNA和tDNA分别产生的各种突变类型的占比。SNV、截断变异和剪接变异主要由ctDNA贡献(分别比tDNA高50%、75%和60%),而CNV(主要是扩增)主要在tDNA中观察到(比ctDNA高70%)。在配对样本中,ctDNA和tDNA对移码事件的贡献相当。
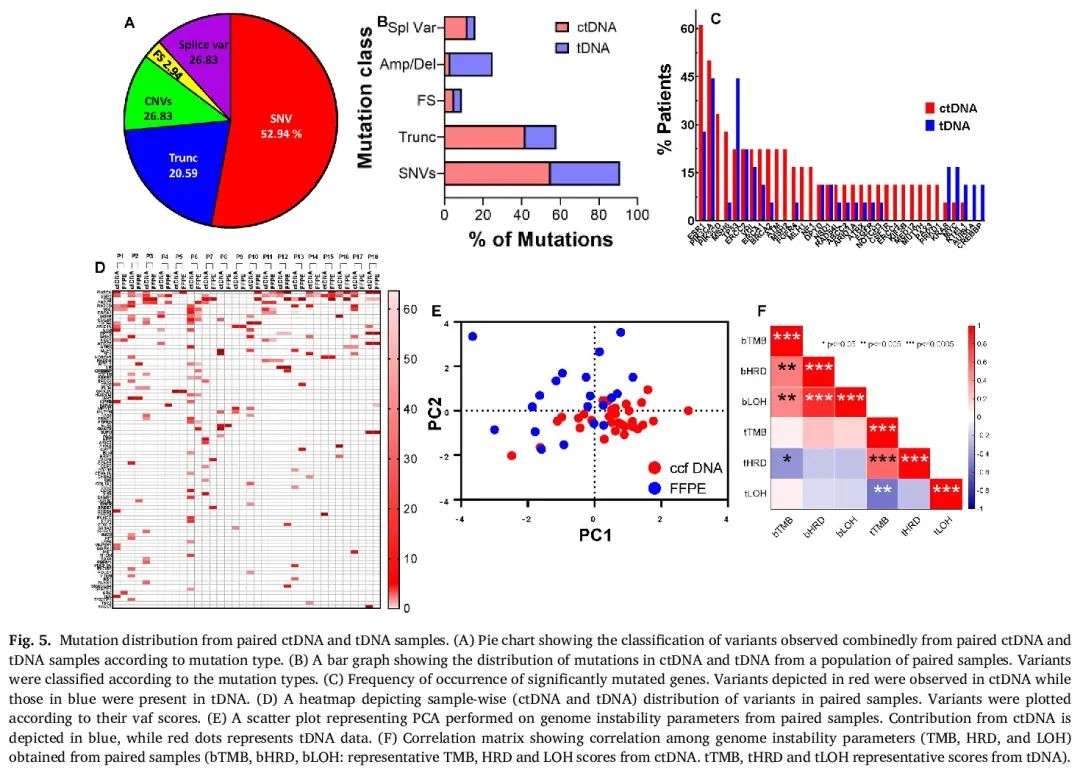
图5
我们观察到,在配对样本中至少存在一种临床重要(致病性或可能致病性)变异的一致性为83.33%,而在配对样本中至少检测到两种变异的一致性为66.66%。在至少有三个突变的配对样本中,22%显示出完全一致性。图5C展示了配对样本中个体变异的分布情况。在个体突变水平上进行衡量时,PIK3CA一致性最高,在39%的配对ctDNA和tDNA样本中检测到。其次是TP53和ESR1,它们在22.22%的配对样本中具有一致性。令人惊讶的是,TP53在tDNA中的频率高于ctDNA。这可能提示携带TP53的肿瘤细胞从原发性肿瘤部位向血液中释放的倾向较低。实际上,在TP53突变主要见于tDNA而未见于ctDNA的配对样本中,我们观察到ctDNA浓度较低。在配对样本队列中,对于所有携带TP53突变的tDNA样本,我们观察到热点PIK3CA变异(E542K、545K或H1047R)作为伴随突变(图5D)。共存的TP53和PIK3CA变异具有临床意义,因为其存在与晚期癌症患者的生存指标相关。
为了确定基因组不稳定性参数之间的关联程度,我们使用TMB、HRD、LOH、LST和TAI评分对ctDNA和tDNA样本进行了主成分分析。图5E散点图直观呈现了主成分分析的结果,提示ctDNA和tDNA之间明显的区分。ctDNA数据主要集中在PC1为正、PC2为负的象限,提示PC1是ctDNA数据集的主要驱动因素。ctDNA样本主要沿PC2与组织样本分离,提示ctDNA存在一种tDNA所没有的独特变异来源。tDNA的参数在PC1和PC2为正的象限中分布较为广泛,提示tDNA中的变异更为复杂,无法用单一主成分解释。由于PC1和PC2都对tDNA变异有贡献,可能存在多个变量或变异模式导致组织样本之间的差异。总体而言,ctDNA和tDNA数据分布的差异表明这两种样本类型可能具有不同的生物学特征,这些差异在NGS分析中有所体现。然而,需要进一步研究以确定HRD和LOH评分是如何导致这些差异的。从主成分分析中获得的信息与基于基因组不稳定性参数的相关矩阵结果相符(图5F)。相关性分析表明,ctDNA样本的基因组不稳定性参数之间存在强相关性(P≤0.005或≤0.0005),而tDNA样本的参数之间既有正相关也有负相关。
我们进行了配对分析,以确定匹配样本的ctDNA和tDNA谱在基因组上是否存在差异,以及这些样本中突变总数和类别的相关性(图6A - F)。在基于突变总数进行比较时,在匹配的ctDNA和tDNA群体中,ctDNA样本中SNV和截断变异的存在显著富集(总突变:P=0.0007,r=0.7;SNV:P=0.0012,r=0.6;截断变异:P<0.0001,r=0.8)。CNV在匹配的ctDNA和tDNA样本之间呈负相关,提示它们主要存在于tDNA样本中。尽管剪接变异具有正相关性(P=0.044,r=0.5),但相关性较弱,提示它们在匹配样本中对ctDNA和tDNA的贡献相似。在个体突变层面,我们观察到许多变异存在相互包含和相互排斥的关系。PIK3CA与神经源位点Notch同源蛋白3(NOTCH3)、TP53和ERBB家族(EGFR和ErbB2)基因突变相互排斥,与NSD1、MSH2和成纤维细胞生长因子受体4(FGFR4)有强关联。同样,ESR1与NOTCH3和ErbB家族基因突变相互排斥,与ATRX突变有强相互包含关系。FGFR4和NOTCH3之间,以及ATM和ERCC2突变之间的相互排斥性最强。另一方面,观察到VHL和ERCC2突变之间有强关联,其次是FGFR4和MSH6。此外,我们注意到无义突变(在ctDNA样本中显著富集)与MMR通路突变之间存在强关联(F检验,P<0.0001,优势比=4.935)。这可能是因为ctDNA比tDNA更有效地捕获肿瘤异质性。
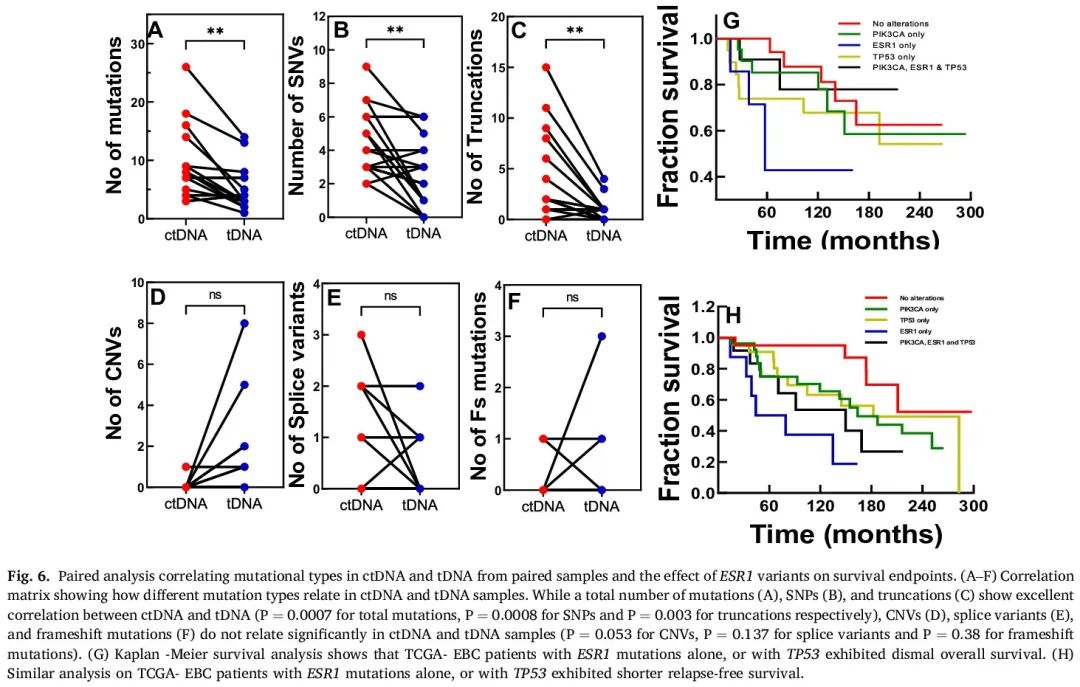
图6
基因组特征与生存终点的相关性
由于我们的数据集显示ESR1变异富集,并且在ctDNA以及配对样本中,它与PIK3CA和TP53突变共存,我们从原发性乳腺癌的癌症基因组图谱(TCGA)数据集中,比较了携带这些突变的患者的总生存期(OS)和无复发生存期(RFS)。如图6G所示,仅携带ESR1突变的患者OS和RFS较差,提示ESR1变异可能是乳腺癌临床预后不良的独立因素。与没有ESR1突变的患者(OS未确定;≥150个月,P=0.015)相比,携带ESR1突变的患者中位OS显著更低(61个月)。此外,ESR1突变与PIK3CA和TP53突变的相关性,在已知转移性乳腺癌患者中更为明显。相应地,生存分析表明,同时携带ESR1、PIK3CA和TP53变异的患者OS较差。这表明原发性肿瘤中同时存在的ESR1、PIK3CA和TP53变异,可能进展,导致激素治疗耐药。实际上,我们观察到同时存在PIK3CA和ESR1,以及TP53和ESR1突变的患者在接受初始根治性标准治疗(手术加化疗)后的24个月内出现转移性复发(代表性病例,显示肿瘤复发和肝转移)。没有ESR1突变(单独或共突变)的患者,无复发生存期较长,且没有临床转移迹象。所有其余患者的临床随访正在进行中,后续将报告结果。
为了进一步评估我们队列中ESR1突变的存在对疾病复发的影响,我们分析了携带ESR1、PIK3CA和TP53突变的EBC患者的TCGA数据。图6H比较了携带和未携带ESR1、PIK3CA和TP53突变的患者的RFS。携带ESR1变异的患者相比未携带ESR1或PIK3CA变异的患者,ESR1的存在显著缩短了患者的无复发生存期(中位RFS为36个月,而未携带ESR1变异的患者未确定,P=0.0033)。PIK3CA和TP53突变对OS和复发持续时间的影响,与ESR1相比没有那么显著。有趣的是,在这些队列中,ESR1突变仅在2.2%的患者中出现,而在我们的研究中(ctDNA和tDNA合并)这一比例为37.5%。这些数据强烈表明,即使在早期,亚克隆ESR1变异的存在,也可能是疾病侵袭性肿瘤生物学行为的强预测指标。
ctDNA图谱中富含与治疗耐药和迁移相关的通路
为了确定EBC队列中观察到的变异在功能层面可能产生的影响,我们对患者进行了KEGG和基因本体定义的通路富集分析。基于在ctDNA和tDNA样本中观察到的总体突变,并考虑共同(共36个共同基因)、ctDNA特有(ctDNA特有的27个基因)和tDNA特有(tDNA特有的21个基因)基因,该分析导致46条乳腺癌相关通路富集。与DNA复制和修复、细胞生长以及信号转导相关的通路,在两个基因集中均富集。有趣的是,与细胞迁移相关的过程(上皮-间质转化、细胞黏附、细胞外基质调节、细胞骨架重塑等),在ctDNA基因集中更为富集。此外,ctDNA样本显示出类似的耐药相关通路富集。另一方面,tDNA基因集中与细胞凋亡相关的通路显著富集。这些数据提示,与tDNA相比,ctDNA分析能够更全面地了解肿瘤突变谱,信息损失最小。
讨 论
研究病理突变的发生情况,有助于从基因组层面深入了解疾病进展机制,为临床提供参考。乳腺癌的转移和复发,是由于对治疗产生获得性耐药,或肿瘤遗传组成相关的生物学异质性所导致。虽然基于组织的NGS分析是检测可干预突变的常规方法,但 “组织是问题”这一共识的兴起,使得基于ctDNA的液体活检在乳腺癌管理中的应用日益广泛。
在本研究中,我们评估了OncoIndx(一种全面的靶向基因组panel),用于分析40例EBC患者的55份ctDNA和tDNA样本的全基因组突变特征。本研究的主要结论是,ctDNA呈现出与tDNA互补但更广泛的突变谱。该突变谱中,循环ESR1、NF1、ARID1A和CCND1/2变异显著富集,这些变异是转移性和复发性肿瘤的特征。与TCGA数据相比,这些变异在EBC队列中并不常见,但在转移性样本中频繁出现。循环ESR1变异的高发生率,可能与印度患者独特的癌症基因型有关。也可能是由于OncoIndx对ESR1基因的高覆盖度和深度测序(5000X),相比以往研究提高了检测灵敏度。同样,ctDNA样本中更频繁地检测到提示与疾病进展(RTK和DDR类共突变)以及治疗耐药相关的突变(ESR1、NF1、ARID1A或ARID1B)(F检验,P=0.03,置信区间:1.1-12.42,错误发现率(FDR)=0.18)。
OncoIndx能够可靠地解读TMB、HRD和LOH评分。一个或多个MMR或HRR基因突变的存在,是衡量基因组不稳定性的指标。我们发现30%的患者携带可能影响MMR通路的变异。在高HRD评分的患者中,82%至少携带一个HRR通路突变,提示HRR突变与HRD评分之间存在良好的相关性(P=0.022,优势比=10.2,FDR=0.1)。基于HRD和TMB评分进行比较时,我们未观察到显著相关性(P=0.9),提示HRD和TMB相互独立,分别代表不同的生物学过程。TMB和HRD评分是治疗决策的重要参数,因为HRR缺陷型肿瘤患者,可能从聚ADP核糖聚合酶抑制剂(PARPi)治疗中获益。除了PD/PD-L1表达外,TMB也是免疫治疗(ICI)的新兴生物标志物。此外,多项研究表明,进展性乳腺癌患者基于TMB评分接受ICI治疗,疗效较好。令人惊讶的是,尽管本研究中的患者处于疾病早期,但仍有约25%的患者具有高TMB(bTMB高:25%;tTMB高:35%)。由于TMB用于免疫治疗决策,与肿瘤的微卫星状态无关,这些患者可能从ICI治疗中获益。本研究中观察到的高TMB评分,可能是由于增殖性肿瘤细胞未接受过治疗,未受到选择压力的影响,突变积累率较高。此外,本研究关于TMB的结论基于较小的样本量,这是本研究的明显局限性。
高TMB评分的肿瘤至少携带一个增殖信号通路突变(P=0.041,优势比=6.5,FDR=0.16,F检验),这强烈表明,在HR+ EBC中,快速增殖的细胞可能积累更多突变,可能从联合治疗中获益。这一观察结果,与之前在乳腺癌、肺癌和结直肠癌中的研究结果一致。考虑到一些携带RTK-PIK3-AKT轴或雌激素通路突变的患者具有较高的HRD评分,他们可能从PARPi-铂类联合阿培利司治疗中获益。同样,高TMB评分且存在增殖信号通路突变的患者,可能从联合ICI治疗中获益。
我们进一步评估了全基因组突变特征的分布,以更好地理解驱动肿瘤进展的突变过程。与既往研究一致,我们发现PIK3通路突变最为常见。除了经典的热点变异(p.E542K、P.E545K和p.H1047R),我们还检测到罕见的循环PIK3CA突变。这些不常见的变异(PIK3CA:p.E81K、PIK3CA:p.Q546H、PIK3CA:p.G1049R)提示螺旋结构域和激酶结构域发生了改变,可能在EBC中导致功能获得。其中一些突变(例如PIK3CA:p.V730I),在晚期乳腺癌组织中出现频率较低,在EBC中尚未作为循环变异被报道。
我们最重要的发现,是研究人群中ESR1变异的高发生率。ESR1变异在转移性和复发性乳腺癌中较为常见,其基础表达与治疗反应密切相关。适应性ESR1突变,在EBC中很少见,而在复发或转移进展时经常获得,导致对内分泌治疗耐药。我们在34%的人群中检测到循环ESR1变异,而其在tDNA中的发生率较低。这一观察结果,与之前一项对比乳腺癌患者组织活检和液体活检的真实世界数据集的研究结果一致。重要的是,90%未接受过化疗的患者中检测到ESR1变异,提示可能的微转移迹象,这与这些患者的组织病理学分析结果相反。这些患者循环肿瘤细胞数量较多(数据未显示),进一步支持了这一结果。根据VAF评分(0.5 - 2%),所有检测到的ESR1突变似乎都是亚克隆性的。考虑到ESR1变异在导致靶向治疗和放射治疗耐药,以及转移进展中的作用,这是一个重要的发现。此外,ESR1变异正成为预测乳腺癌亚型转换的重要标志物,因为治疗反应显著受到乳腺癌亚型的影响。与ESR1野生型肿瘤患者相比,同时存在ESR1和PIK3CA或TP53突变的患者,出现迅速复发和肝转移。
除了PIK3CA和ESR1,血浆TP53和BRCA变异分别见于28%和25%的人群,而在tDNA样本中,这些变异的检出率分别为37%和23%。这一观察结果与既往研究一致。HRR通路和PIK3CA信号通路突变似乎相互关联,在所有携带PIK3CA突变的ctDNA样本中同时存在,提示两者之间存在因果关系(Spearman相关系数R=0.652,P=0.009)。同样,在配对样本中,34%的ctDNA和11%的tDNA中,PIK3CA和ESR1变异同时出现。除了BRCA,我们还观察到9个直接影响HRR通路的变异,提示DDR和同源重组功能的显著失调。
此外,我们进行了通路富集分析,以进一步了解突变谱如何影响肿瘤细胞的功能行为。通路分析识别了多个KEGG定义的显著失调的通路。特别值得关注的是,与耐药和迁移表型相关的通路富集,因为它们提示侵袭性疾病和化疗耐药。尽管本研究表明,EBC中与转移和治疗耐药相关的突变富集,但研究仅限于单一时间点一小部分患者。此外,由于本研究为回顾性研究,未纳入临床评估指标,因此无法将观察到的突变谱与生存结局直接关联。另外,我们的研究仅限于靶向基因组区域,未考虑非编码变异、转录和表观遗传失调,以及与表型标志物的相关性。
本研究通过液体活检,对EBC进行了全面的基因组分析,揭示了ctDNA和tDNA之间显著的突变异质性。ctDNA呈现出更广泛、更丰富的突变图谱,尤其是在与转移和治疗耐药密切相关的基因中。值得注意的是,ESR1变异在未接受化疗的患者中高度存在,提示传统基于小基因panel和低测序深度的NGS方法,可能会忽略潜在的微转移活动。尽管本研究受到回顾性研究设计、样本量较小和缺乏生存数据的局限性,但它为了解驱动肿瘤进展和治疗耐药的分子机制,提供了重要信息。这些发现,为临床试验更精确地进行患者分层,以及开发个性化治疗方案,奠定了基础。未来的研究应拓展这些研究成果,纳入更大的患者队列,并确定与患者生存结局的相关性。
参考文献:
Atul Bharde, Snigdha Nadagouda, Manoj Dongare, Kanchan Hariramani, Madhura Basavalingegowda, Sumit Haldar, Alain D'Souza, Bhagwat Jadhav, Sangeeta Prajapati, Vikas Jadhav, Sujit Joshi, Aravindan Vasudevan, Mohan Uttarwar, Wenhui Zhou, Sirish Kishore, Kumar Prabhash, Jayant Khandare, Gowhar Shafi, ctDNA-based liquid biopsy reveals wider mutational profile with therapy resistance and metastasis susceptibility signatures in early-stage breast cancer patients, The Journal of Liquid Biopsy, Volume 7, 2025, 100284, ISSN 2950-1954, https://doi.org/10.1016/j.jlb.2024.100284.


