NBT:生物信息平台DRAGEN可30分钟完成大规模全基因组分析和多种变异检测
时间:2024-11-14 12:02:02 热度:37.1℃ 作者:网络
近年来,越来越多的研究使用全基因组测序(WGS)解析变异对整个基因组的影响。精确检测单核苷酸变异(SNV)和小片段插入或缺失(Indel)的方法一直处于被研究的前沿,结构变异(SV)和拷贝数变异(CNV)也被纳入了分析。另一种未被充分研究的变异类型是短串联重复序列(STR)扩增。所有这些变异类型都存在于整个基因组中,但由于联合检测和准确报告变异类别的挑战,许多研究往往只关注SNV或独立的变异类型子集。此外,来自基因组高度多样化和重复的区域,使分析更加复杂。以上变异类型可能会相互作用,但在单独分析时,这些关系就会丢失,进一步增加了重复片段所带来的检测挑战。因此,目前仍然缺乏可扩展的、统一的软件框架来全面识别所有变异类型。
近日,美国贝勒医学院、Illumina公司等单位的研究人员在Nature Biotechnology发表了题为“Comprehensive genome analysis and variant detection at scale using DRAGEN”的文章,报道了基因组学的动态读取分析方法(Dynamic read analysis for genomics,DRAGEN)在WGS平台上大规模地综合鉴定多种胚系变异类型的能力。DRAGEN基于泛基因组参考的多基因组映射(提高准确性)、硬件加速和基于机器学习的变异检测来提供对整个基因组变异的见解,从原始读取到变异检测的计算时间约为30分钟,且在所有变异类型(SNV、Indel、STR、SV和CNV)检测的速度和准确性方面优于当前最先进的方法。研究团队通过分析1000基因组计划(1kGP)说明了DRAGEN在大型队列中的扩展能力。总体而言,DRAGEN是一种高精度、快速和可扩展的方法,标志着测序数据分析的一个重要里程碑,将通过一个高度全面和可扩展的平台,为发现孟德尔病和罕见病等各种疾病相关的准确和潜在变异提供新见解。

主要研究内容
DRAGEN(v.4.2.4)可用于识别所有类型的基因组变异(图1):首先,每个样本被映射到一个泛基因组参考(由一个参照和几个装配体组成),以克服人类特定参考基因组的错误,泛基因组参考包括来自多个基因组组合的变异,以更好地代表整个人类群体中个体之间的序列多样性,并且可以通过数据库更新实现更精确和全面的短读长测序数据比对。为了识别SNV和Indel, DRAGEN使用de Bruijn图组装具有变异的区域,然后将其输入到隐马尔可夫模型中,该模型具有先前估计的每个样本的噪声和误差水平,输出为VCF文件。在初始变量调用之后,机器学习框架会重新调用,以进一步减少假阳性的小变异(SNV和Indel),并恢复错误丢弃的假阴性。同时,DRAGEN使用两种方法识别SV和CNV。对于SV调用,DRAGEN通过引入一些关键概念扩展了Manta,大大改善了SV调用;对于CNV调用,DRAGEN靶向导致基因组片段扩增或删除的1kb和更大的变异,还被设计考虑来自SV调用的不一致和分裂读取信号,以检测低至1kbp的事件。此外,DRAGEN还可识别STR,并使用主要基于ExpansionHunter的方法分析已知的致病基因组区域。
一些重要基因由于其序列与假基因高度相似、重复区域和多态性,对基因分型具有挑战性。为了克服这些挑战,DRAGEN整合了9个靶向调用基因,对临床相关基因(CYP2B6、CYP2D6、CYP21A2、GBA、HBA、LPA、RH、SMN和HLA)进行准确的基因分型,其中6个在该文章中进行了描述。
分析显示,DRAGEN可以在大约30分钟内处理35x全人类基因组Illumina fastq文件,对SNV、Indel、STR、SV和CNV进行全基因组同步评估,并报告来自靶向调用基因的结果。因此,DRAGEN能够在整个基因组中大规模捕获单个变异和较大变异,并在标准化的VCF文件中报告,以人类基因组中大规模地生成一套全面而准确的基因组变异图谱。
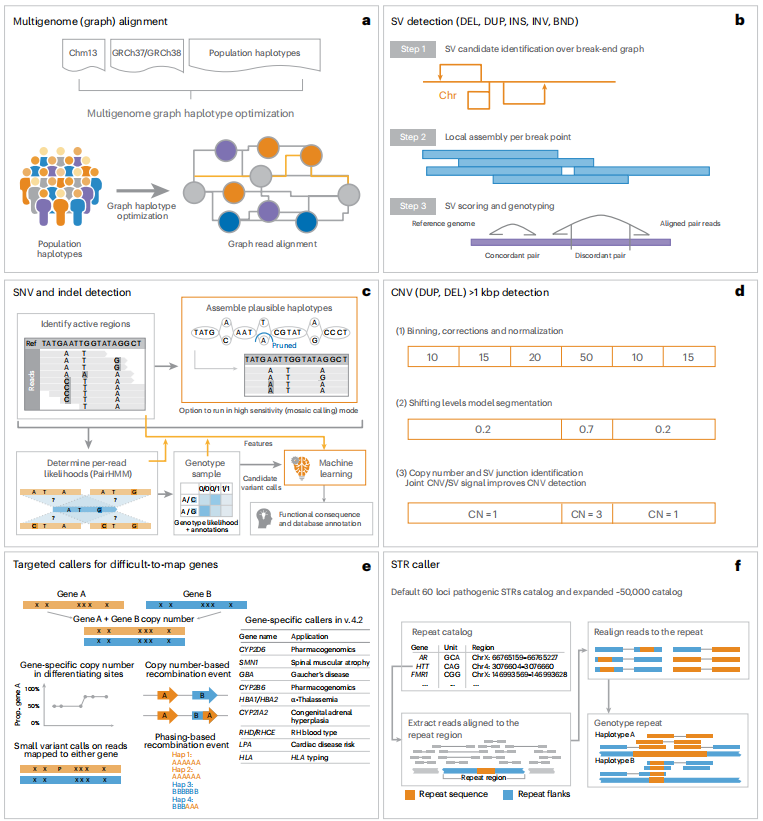
图1. DRAGEN分析流程概述
研究人员将DRAGEN应用于HG002样本,并对检测到的所有大小的变异分布进行分析,突出显示了DRAGEN捕获整个变异谱的能力。结果发现,DRAGEN捕获了492万个小变异(包括3,956,307个SNV),960908个Indel,13886个SV(≥50 bp),1156个CNV(1 kb-445 kb)以及50069个STR目录中的31370个STR扩增或收缩多态性位点。更加重要的是,在所有基准测试中,DRAGEN显示出更高的准确性和令人印象深刻的从原始读取到最终变异调用的分析速度,总共在30分钟内,这比任何其他现有的工作流程都要好。与其他最先进的方法相比,研究团队证明了DRAGEN在SVs、CNVs和STR发现方面的重大改进,这突出表明虽然等位基因信号存在于短读长测序数据的复杂等位基因中,但需要更先进的方法来准确地破译和报告它们。
整体来说,DRAGEN的优良性能在一定程度上是通过利用具有泛基因组参考的多基因组制图实现的。最新发布的DRAGEN版本包括一个64单倍型泛基因组参考,代表了不同的人类祖先,随着更多的泛基因组样本的增加,参考基因组也会随之更新。分析数据表明,使用当前的64单倍型泛基因组参考,DRAGEN不仅在准确性上优于现有的泛基因组分析方法,而且在可扩展性和运行时间上也优于其他。此外,DRAGEN泛基因组分析也纳入了SV和CNV检测,这是目前其他泛基因组分析工具无法实现的。

图2. DRAGEN性能概述
为了进一步提高DRAGEN在种群水平上的可扩展性,研究团队还提出了提供种群水平VCF文件的方法,这些文件是任何后续全基因组关联研究或其他功能研究所必需的。研究提出的DRAGEN迭代VCF基因型,可以获得完全基因分型的多样本VCF文件,并且分析结果显示其可识别许多以前未报道的变异,这些变异不仅在基因组中,还包括在重要的医学相关基因中。此外,研究人员还克服了在个体和群体水平上整合STR、SV和CNV的挑战。对于1kGP队列数据集,DRAGEN能够发现更多的变异,即SNV、Indel和大片段变异,除了这些变异外,DRAGEN还在基因组中发现了STR扩增和CNV。此外,研究还展示了DRAGEN在3,202个全基因组测序数据集上的性能,并展示了其可扩展性、准确性和创新性。
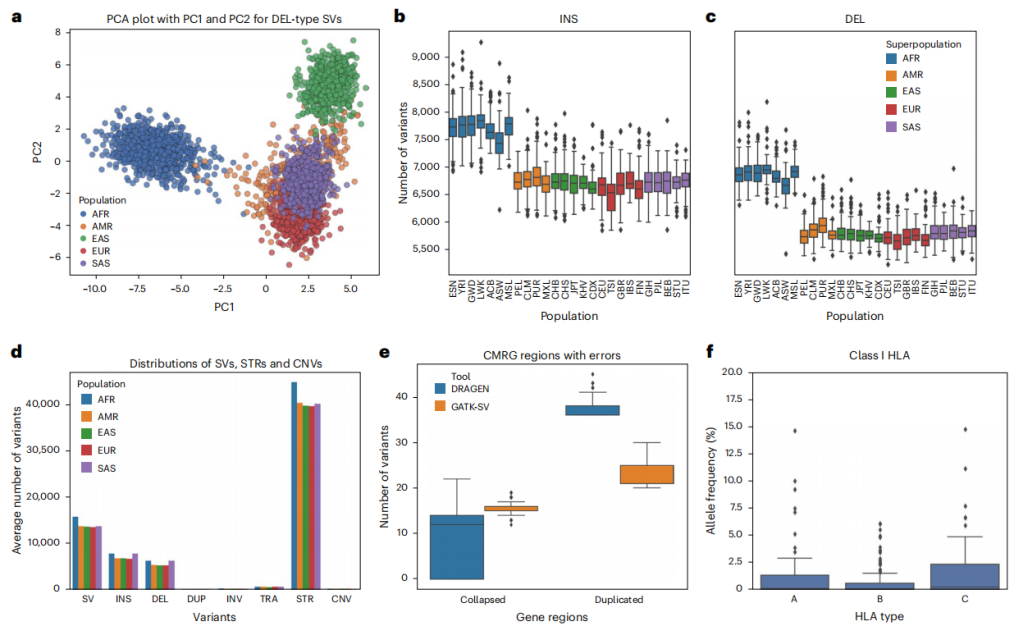
图3. DRAGEN在1kGP队列数据集上的应用
结 语
该研究介绍了DRAGEN基本架构及其在SNV和Indel调用中的优异性能,以及它检测整个变异图谱的能力。DRAGEN代表了基因组算法发展的进步,以解决在所有类型等位基因中检测变异的可扩展性、准确性和全局性的长期问题,从而在规模上经济有效地全面解析整个基因组。
研究团队引入并测试了DRAGEN的各项分析模块,并通过分析1kGP队列数据集说明了其在大型队列中的扩展能力。同时,研究还揭示了跨人群基因组多样性的新见解,并特别关注了医学相关基因,以展示DRAGEN的基因组和医学效用。此外,DRAGEN纳入了特定方法来比较和合并产生的变异,以进一步强调DRAGEN分析多个变体类的能力。因此,DRAGEN能够以前所未有的规模和准确性评估变异,为医学和生物学研究提供新的见解。目前,DRAGEN直接集成到Illumina测序仪中,已经被部署在几个大型项目中,比如UK Biobank和All of Us等等。
DRAGEN代表了测序数据分析的一个重要里程碑,并将作为一个高度全面和可扩展的重要平台为导致许多疾病的机理提供新见解。
论文原文:
Behera, S., Catreux, S., Rossi, M. et al. Comprehensive genome analysis and variant detection at scale using DRAGEN. Nat Biotechnol (2024).
https://www.nature.com/articles/s41587-024-02382-1

