神经内分泌肿瘤患者检出MEN1 c.587delA胚系杂合变异,确诊家族性多发性内分泌腺瘤病1型
时间:2024-12-22 14:00:40 热度:37.1℃ 作者:网络
多发性内分泌腺瘤病1型(MEN1)是一种罕见的遗传病,其特征是数种内分泌系统病变同时发生。在MEN1中,MEN1基因致病突变导致关键的肿瘤抑制蛋白menin异常表达。本文报告了一例患有胰岛细胞瘤和原发性甲状旁腺功能亢进症的 14 岁男性病例。基因检测显示MEN1基因存在一种新的杂合变异c.587delA,导致第 196 位氨基酸被替换,从谷氨酸变为甘氨酸,随后 33 个氨基酸发生移码翻译。在先证者(即患者)的父亲身上也发现了相同变异——同样进一步诊断为甲状旁腺功能亢进症。据研究人员所知,这是首例由MEN1 c.587delA变异导致的MEN1综合征的报道。观察表明,尽管携带相同的MEN1基因变异,但家族成员表现出不同的临床表型。这强调了家族背景下遗传早现(genetic anticipation)的存在。
背 景
多发性内分泌腺瘤病1型(MEN1)是一种罕见的遗传病,其特征是内分泌系统的几种病变同时发生。MEN1 的患病率为 3-10/100,000。MEN1最常累及甲状旁腺和胰腺、垂体、肾上腺、肺和胸腺,按发病率降序排列。渗透程度还取决于受影响的具体内分泌器官。该病的外显率随着年龄的增长而增加,最早在 10 岁开始,到 60 岁时达到接近普遍水平。该疾病的传播方式是常染色体显性遗传,与位于11q13的MEN1基因杂合失活变异有关。MEN1基因长 9 千碱基,有 10 个外显子,编码蛋白menin,该蛋白由 610 个氨基酸组成,通过与众多伴侣的相互作用在各种细胞机制中发挥关键作用。MEN1是一种抑癌基因,该基因变异通过Knudson提出的“双打击”假说导致神经内分泌肿瘤(NET)。
根据 2012 年制定的诊断标准,MEN1可通过以下三项标准之一进行诊断:①临床标准,存在至少 2 处主要MEN1病变;②家族标准,表现为临床MEN1患者的一级亲属存在MEN1病变;③遗传标准,患者存在MEN1基因的致病性或可能致病性变异,无论是否有症状。根据标准表现为临床MEN1或疑似MEN1的患者应接受基因检测以筛查MEN1基因异常。任何由此识别出的MEN1基因致病性或可能致病性变异都必须在第二个样本上进行检查。临床诊断为MEN1的患者的家庭成员患有MEN1相关肿瘤,则符合家族性MEN1诊断的标准。
本文描述了一个因MEN1新型杂合致病变异c.587delA而患有MEN1的家族。该变异导致第 196 个氨基酸被替换,从谷氨酸变为甘氨酸,随后移码翻译 33 个氨基酸,然后提前终止。此外,本病例还强调了携带相同基因变异的家庭成员之间疾病表型的多样性。
病 例
患者男,14 岁,反复癫痫样发作 2 年多,发作停止后意识不清,空腹血糖 2.09 mmol/L(正常范围 3.9-5.9),胰岛素 23.96 μU/ml(正常范围 1.5-15),C肽 0.972 nmol/L(正常范围 0.48-0.78)。增强腹部磁共振成像(MRI)扫描提示胰体前部有结节性病变,提示为神经内分泌肿瘤(图1)。入院体格检查未发现任何异常。
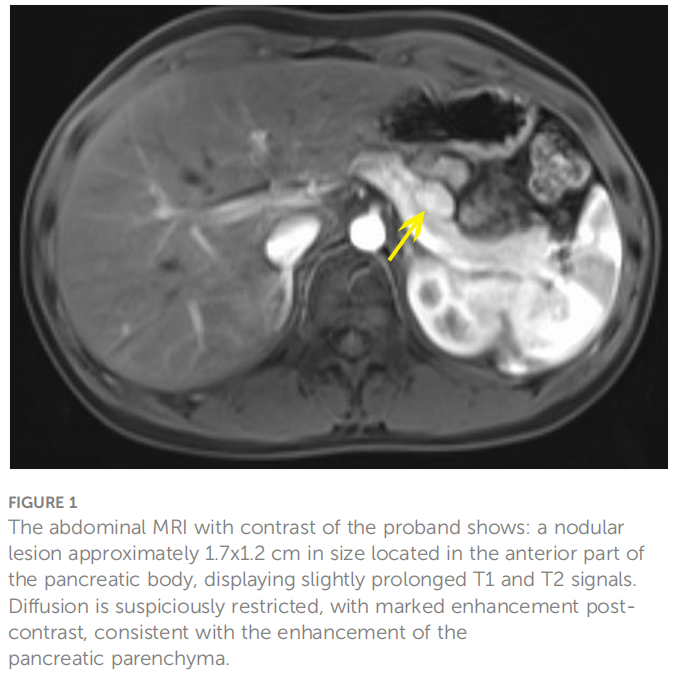
▲图1 先证者腹部增强MRI检查示:胰体前部有一结节性病变,大小约1.7x1.2cm,T1、T2信号稍延长,弥散受限可疑,增强后强化明显,与胰腺实质强化一致
2019 年 1 月 29 日患者在全身麻醉下行远端胰腺切除术及胰腺部分切除术。胰腺病理报告肿瘤细胞阳性PCK(点状+)、CgA(+)、Syn(+)、CD56(+)、ATRX(+)、Rb(散发+)、P53(+,5%),Ki-67阳性率为 5%-10%,诊断为 2 级神经内分泌肿瘤(NET G2)。免疫组化显示胰岛素阳性,胰高血糖素、胃泌素、生长抑素阴性,最终诊断为胰岛细胞瘤。术后空腹血糖 4.28-5.02 mmol/L,空腹胰岛素 6.28-13.18 μU/ml,无术后癫痫样发作。住院期间钙、磷、甲状旁腺激素水平正常。但随访期间血钙、甲状旁腺激素水平升高,磷水平降低。监测血钙水平为 2.62-2.81 mmol/L(正常范围2.1-2.7 mmol/L),磷为1.07-1.33 mmol/L(正常范围 0.81-1.45 mmol/L),甲状旁腺激素为 9.85-11.35 pmol/L(正常范围 1.60-6.90 pmol/L),24 小时尿钙排泄率为 12.94 mmol/24h(正常范围 2.5-7.5),尿磷为 27.0 mmol/24h(正常范围 22-48)。骨代谢标志物提示骨转换明显增加。患者无多饮、多尿、恶心、呕吐、腹痛、腰痛、血尿、骨痛等症状。甲状旁腺超声示:双侧甲状旁腺深部实性结节。甲状旁腺单光子发射计算机断层扫描/计算机断层扫描(SPECT/CT)显示:4 个甲状旁腺均能显影,甲状旁腺锝-99m甲氧基异丁基异腈(99mTc-MIBI)扫描摄取无明显增加(图2)。
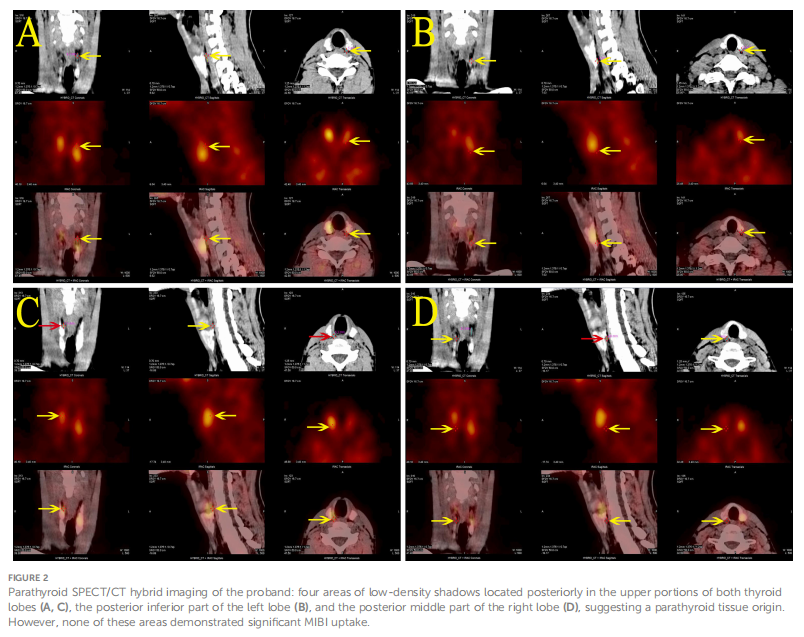
▲图2 患者甲状旁腺SPECT/CT混合显像
骨密度属同龄正常范围。泌尿系统超声未见泌尿道结石。2019 年 7 月 3 日患者在全麻状态下行“颈部探查+甲状旁腺全切除+甲状旁腺部分自体移植术”。左上、左下、右上甲状旁腺组织石蜡病理诊断提示增生。手术成功,术前测PTH 11.44 pmol/L,术后 30 min测PTH 1.15 pmol/L。术后治疗包括补钙、维生素D,术后 4 周PTH 1.98 pmol/L,血钙 2.33 mmol/L,磷酸盐 1.7 mmol/L。逐渐停用药物,迄今为止的随访显示血钙、磷酸盐、甲状旁腺激素水平正常,无低血糖或癫痫发作,也没有其它MEN1相关全身性疾病的症状或生化标志物。考虑到患者同时存在胰岛细胞瘤和甲状旁腺功能亢进,研究人员对其进行了MEN1相关疾病表型筛查和基因检测。患者的胃泌素水平、垂体及靶腺轴激素、血尿儿茶酚胺及其代谢物、醛固酮-肾素-血管紧张素系统、全血细胞计数、肝肾功能、尿液分析和肿瘤标志物(AFP、CEA、CA19-9、CYFRA21-1、NSE)均在正常范围内。肾脏、肾上腺、阴囊超声检查以及胸部和头部CT扫描均未见异常。垂体MRI未发现肿块病变。对该患者进行深度全外显子组测序,在 11 号染色体(chr11:64575445)上发现一个遗传变异MEN1 c.587delA(图3A),导致氨基酸序列改变为p.Glu196GlyfsTer33(NM_130803)。这表明编码区第 587 位发生碱基A缺失,导致第 196 位氨基酸由谷氨酸替换为甘氨酸,随后发生 33 个氨基酸的移码翻译,随后翻译终止。总测序深度为 144,变异深度为 84,表明为杂合性。根据 2015 年ACMG指南,该变异被归类为可能致病:PVS1(功能丧失变异可能导致基因失活)+ PM2(MAF <0.005,被认为是低频变异)。
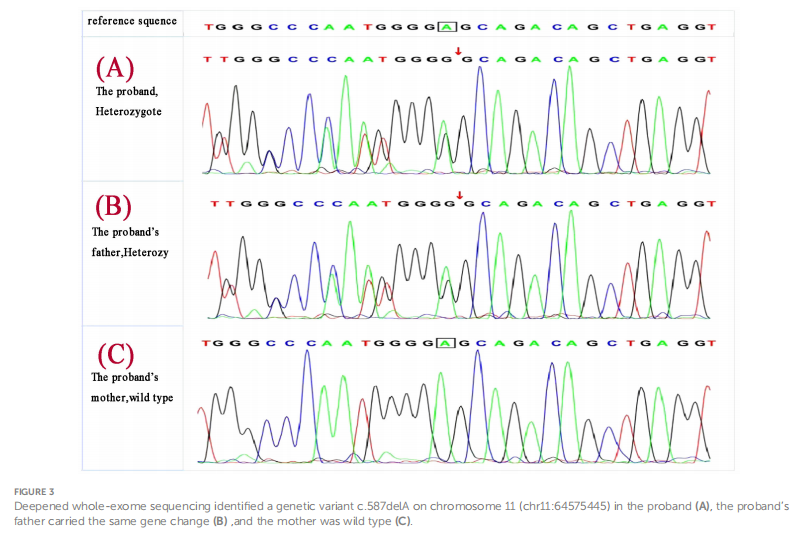
▲图3 (A)深度全外显子组测序发现先证者第11号染色体(chr11:64575445)上存在一个遗传变异c.587delA;(B)先证者父亲携带相同的基因变异;(C)母亲为野生型
患者父母均进行了MEN1基因检测,父亲携带相同基因变异,亦为杂合子(图3B),母亲为野生型(图3C)。患者父亲,男性,43 岁,反复腰痛病史 6 年余,因双侧输尿管结石多次接受手术,包括体外冲击波碎石术、经尿道输尿管切开取石术,术后分析证实结石为草酸钙。体格检查无阳性发现。进一步的检查显示甲状旁腺激素(PTH)为 24.28 pmol/L,血钙为 2.77 mmol/L,血清无机磷为 0.67 mmol/L,空腹血糖为 5.67 mmol/L。24 小时尿电解质分析显示钙为 4.38 mmol/24h,磷为 16.20 mmol/24h。骨代谢标志物提示骨转换显著增加。胃泌素、垂体及靶腺轴激素、血和尿儿茶酚胺及其代谢物、醛固酮-肾素-血管紧张素系统、空腹血糖、胰岛素、C肽、全血细胞计数、肝肾功能、尿液分析和肿瘤标志物均正常。胰腺增强MRI和垂体MRI未见明确异常。甲状旁腺SPECT/CT混合显像显示右甲状腺叶下部后方结节MIBI摄取轻度增加,提示甲状旁腺组织来源(图4)。2019 年 7 月 9 日,根据患者父亲不愿行甲状旁腺次全切除术的意愿,实施了颈部探查及右下甲状旁腺腺瘤切除术。病理提示右下甲状旁腺腺瘤样增生,免疫组化示PTH(+)、CD56(-)、Syn(+,部分)、CgA(+),Ki-67阳性率约 1-2%,网状纤维(Foot)特殊染色阳性。术后复查血钙 2.51 mmol/L,磷酸盐 0.95 mmol/L。随访至 2024 年 2 月 18 日,父亲血钙、磷酸盐、PTH均正常,未再发生尿路结石,未出现其它MEN1相关疾病表型。
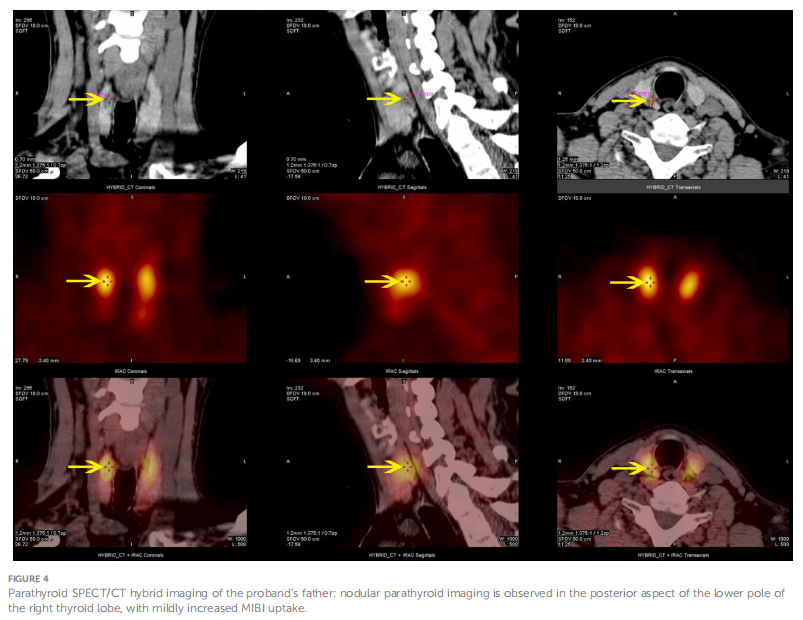
▲图4 先证者父亲甲状旁腺SPECT/CT混合显像
先证者无兄弟姐妹,其祖父母和父亲的兄弟姐妹不愿接受基因检测,仅选择筛查血糖、钙、磷和甲状旁腺激素,结果均正常。本病例的c.587delA基因变异为杂合子,与父亲的表型和基因型共分离,符合常染色体显性遗传。c.587delA变异被确认为导致家族性MEN1的致病基因变异。综上所述,研究人员整理了一个全面的表格,详细列出了先证者及其父亲在整个诊断和治疗过程中的治疗干预和实验室检查结果。
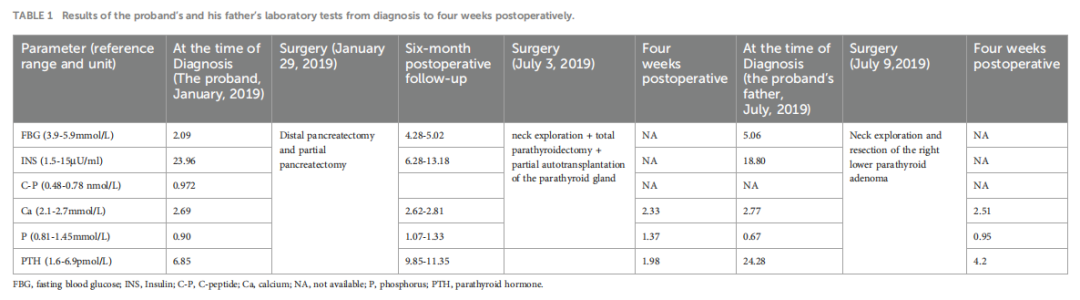
▲表1 患者及其父亲从确诊至术后四周的实验室检查结果
讨 论
MEN1基因编码menin蛋白,该蛋白是一种参与组蛋白修饰和表观遗传基因调控的支架蛋白,在与MEN1相关肿瘤抑制中发挥作用。menin与 50 多种已知蛋白质相互作用,影响各种细胞过程,如细胞周期进程、DNA修复和转录调控。它与转录因子和染色质修饰蛋白相互作用,影响TGF-β/BMP、核受体、Wnt/β-catenin和Hedgehog等对基因表达调控至关重要的通路。menin的缺失会影响这些相互作用,阻碍这些信号通路及其抗增殖作用。基因变异导致的menin失活可使其丧失肿瘤抑制功能。MEN1的致病变异通常会对menin产生截断作用,导致其丧失肿瘤抑制功能并增加罹患癌症的风险。
已发现MEN1基因中 1,300 多种变异,这些变异分布在整个开放阅读框中,主要存在于编码外显子和内含子序列中,没有明显的聚集或明显的热点。MEN1中约 69% 的胚系变异被认为是致病的,导致menin过早截断。这主要是由于移码变异(42%)和无义变异(14%),以及由剪接缺陷(10.5%)和大量缺失(2.5%)导致的外显子区域缺失。
如果认为某种之前未报告过的基因变异具有致病性,则要确认其致病性通常需要在另一位患病的一级亲属身上证明存在相同变异。MEN1的临床诊断依赖于检测至少两个通常受累器官的肿瘤性疾病,例如甲状旁腺、垂体前叶和胰腺。对于本文的年轻男性患者,胰腺是MEN1的最初征兆,随后发现胰岛细胞瘤和原发性甲状旁腺功能亢进症。基因检测发现了一种新的杂合MEN1 c.587delA变异,导致第 196 位氨基酸从谷氨酸替换为甘氨酸,随后 33 个氨基酸发生移码翻译,然后翻译终止。基因检测表明,患者父亲携带相同的变异,进一步确诊患有甲状旁腺功能亢进症。在患者父亲中发现了与其儿子相同的MEN1症状,从而可以在新的背景下验证该变异的致病性。研究人员认为MEN1 c.587delA变异及移码变异导致menin翻译异常,从而损害其肿瘤抑制功能,导致MEN1的发生。
观察结果表明,尽管家庭成员具有相同的MEN1基因变异,但他们表现出不同的临床表型。父亲成年后才发病,表现为单发甲状旁腺腺瘤,而儿子在 12 岁之前因胰岛细胞瘤出现严重的低血糖症状,随后因多个甲状旁腺增生而出现高钙血症。这强调了家族背景中存在遗传早现(genetic anticipation)。此前已记录到发病年龄、临床症状、疾病严重程度和肿瘤类型存在相当大的差异。受影响腺体的表现及其具体病理,如增生或单发或多发甲状旁腺腺瘤,在家庭成员之间(包括同卵双胞胎)可能有所不同。大量研究分析了携带相同变异的患者及其亲属的临床特征,结果证实表型与基因型之间不存在直接相关性。有人提出,环境因素激活的表观遗传机制可能会影响携带相同MEN1变异的个体的表型。因此,临床表型因疾病外显率不同以及基因-环境相互作用可能产生的影响而存在异质性。
在本文案例研究中,对患者及其父母进行了全外显子组测序。遗憾的是,患者的其他家庭成员不愿意接受基因检测,无法构建该疾病的详细家族谱系。研究人员打算对患者、其后代以及亲属进行持续监测,以观察MEN1疾病表型的后续变化。研究人员认为,本文病例代表了一种家族性MEN1,可归因于新的MEN1变异c.587delA.,该变异导致移码翻译和menin异常表达。先证者和他的父亲均携带相同变异,但表现出不同的临床表型,且症状严重程度不同。基因检测对于MEN1患者至关重要,表观遗传机制可能会影响携带相同变体的个体的表型。
参考文献:
Huang H, Li J, Zhang K, Tang Y, Zhang M, Fan Z, Wang T and Liu Y (2024) Case report: Novel germline c.587delA pathogenic variant in familial multiple endocrine neoplasia type 1. Front. Endocrinol. 15:1467882. doi: 10.3389/fendo.2024.1467882


