病例报告|糖原贮积病Ⅱ型1例
时间:2025-01-15 12:15:02 热度:37.1℃ 作者:网络
摘 要 报告1例糖原贮积病Ⅱ型患者。患者为26岁男性,青年期表现为四肢骨骼肌萎缩无力及心肌受累,肌酶、肌电图、下肢磁共振及肌肉病理活检提示肌炎改变,外周血淋巴细胞滤纸片酶学检查显示酸性α-葡萄糖苷酶(acid alpha glucosidase, GAA)活性部分缺乏,最终检测GAA基因确诊为糖原贮积病Ⅱ型,基因变异c.-32-13T>G和c.1551+2T>G分别来自母亲、父亲。分析该患者发病特点,可认识并积累关于糖原贮积病Ⅱ型病例资料,加深对罕见的常染色体隐性遗传病理解。
关键词
骨骼肌萎缩无力;心肌受累;肌炎;酸性α-葡萄糖苷酶;GAA基因;糖原贮积病Ⅱ型;常染色体隐性遗传病
糖原贮积病Ⅱ型(又称庞贝病)是由酸性α-葡萄糖苷酶(acid alpha glucosidase, GAA)缺乏引起的常染色体隐性遗传病。根据酶活性部分或完全缺乏将庞贝病分为早发型及晚发型。晚发型庞贝病中,骨骼肌受累是主要临床表现。生化检查、肌肉活检以及基因检测对该病诊断必不可少。本文报告1例晚发型庞贝病患者,根据基因检测提示GAA基因存在两个致病突变,且该患者的两个致病变异基因尚未在“庞贝病GAA变异数据库”中报告。本病例报告旨在扩充庞贝病数据库,为庞贝病诊疗提供更多临床资料。
1 临床资料
患者,男,26岁,因“进行性四肢肌肉乏力10年余”于2022年7月来诊。患者10余年前无明显诱因逐渐出现四肢肢体乏力及肌肉萎缩,8年前在外院查肌电图示肌源性损害,当时诊断为“肌营养不良”,未进一步诊治。近半年提重物困难,运动时气促,上述症状无晨轻暮重。无呼吸困难,无胸痛,无肌跳、肌痛等。既往史、个人史均无特殊,家族成员无类似症状患者。
一般生命体征检查正常。神清,体型消瘦,慢性病容,皮肤黏膜无黄染、皮疹、出血点等,心、肺、腹查体无特殊。神经系统专科检查:高级神经及颅神经检查未见明确异常;四肢肌肉萎缩,四肢近端肌力5-级,远端肌力5级;四肢肌张力正常;四肢腱反射消失;感觉系统未见异常;双侧病理征阴性,脑膜刺激征阴性。
肌酸激酶1426 U/L(参考值38~174 U/L),谷草转氨酶449 U/L(参考值15~40 U/L),乳酸脱氢酶539 U/L(参考值120~230 U/L)。血乳酸运动试验:安静时1.3 mmol/L(参考值0.5~1.7 mmol/L),爬楼梯5 min时3.3 mmol/L,休息15 min时1.9 mmol/L。血常规、尿便常规、电解质、肾功能、血脂、甲状腺功能无异常。心电图:窦性心律,不完全性右束支传导阻滞,左心室高电压。心脏彩超:二尖瓣前叶脱垂,除左心室外,其余心腔偏小。肌电图:肌源性损害;上下肢周围神经传导速度及F波未见异常。双下肢磁共振检查(图1)可见双侧大腿、小腿肌肉水肿信号,提示肌炎。
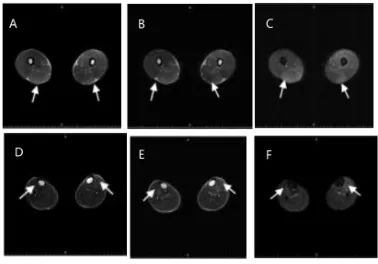
图1 患者双下肢核磁共振 A、B、C可见大腿T1长信号、T2长信号、T2压脂呈高信号水肿病灶,如箭头所示;D、E、F可见小腿T1长信号、T2长信号、T2压脂呈高信号水肿病灶,如箭头所示。Fig.1 MRI of the patient’s lower extremities
病理检查:苏木精-伊红(H-E)染色显示肌纤维轻度大小不等,部分肌纤维见空泡结构,部分空泡见嗜碱性颗粒,考虑为糖原空泡(图2A、B),改良Gomori法(MGT)染色显示糖原空泡嗜碱性颗粒红染(图2C),过碘酸希夫染色(PAS)显示糖原含量增多(图2D),电镜显示糖原沉积(图2E、F)。上述病理改变符合肌源性损害,较符合糖原贮积病Ⅱ型可能。
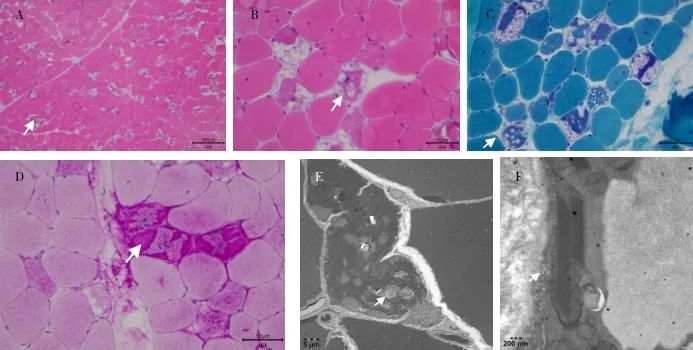
图2 病理检查 A、B为苏木精-伊红(H-E)染色见肌纤维内糖原空泡及空泡内浓染的嗜碱性颗粒,如箭头所示;C为改良Gomori法(MGT)染色显示糖原空泡及空泡边缘红染的嗜碱性颗粒,如箭头所示;D为过碘酸希夫染色(PAS)显示肌纤维内大量的糖原,如箭头所示;E、F为电镜显示空泡及肌原纤维间蓄积大量糖原,如箭头所示。Fig.2 Pathological examination
外周血淋巴细胞滤纸片酶学检查显示酸性α-葡萄糖苷酶活性为0.40 μmol/(L·h)[参考值1.46~20.34 μmol/(L·h)],明显偏低,提示GAA缺乏症。二代测序结果表明GAA 基因上存在c.-32-13T>G和c.1551+2T>G杂合突变(表1),上述变异按美国医学遗传学与基因组学学会(ACMG)《遗传变异分类标准与指南》评级为致病变异[1]。家系验证结果显示此两个杂合突变分别来自于其父母,符合常染色体隐性遗传规律(图3)。因此,根据临床表现、病理改变、酶学检测、基因突变结果,该患者诊断为晚发型庞贝病。现阶段庞贝病有效治疗为重组人GAA的酶替代疗法(enzyme replacement therapy,ERT),但本例患者由于经济原因未进行酶替代治疗。出院随访病情尚稳定,定期在康复医院行运动康复训练以减缓活动耐力下降。虽然文献中只有少数晚发型庞贝病患者出现肥厚型心肌病报道[2],但本例患者心脏彩超已提示部分心腔缩小,随访时建议后期监测心功能变化。
表1 致病性基因变异Tab.1 Pathogenic gene variations

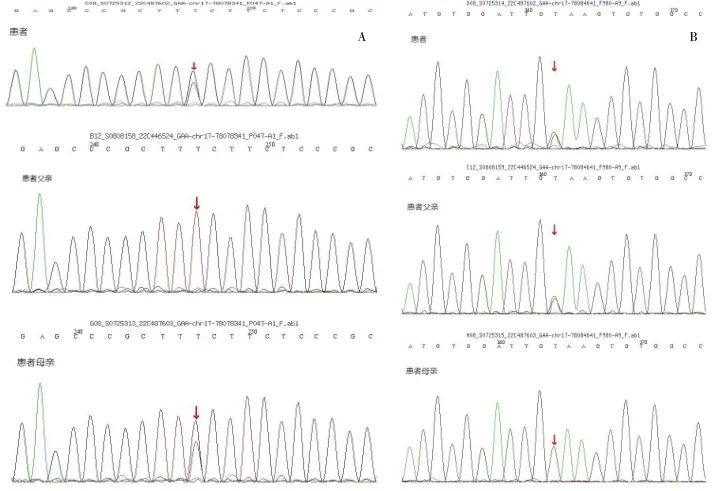
图3 Chr17:78078341和Chr17:78084641 GAA基因测序图 A. Chr17:78078341 GAA基因测序显示,患者杂合突变,患者父亲无变异,患者母亲杂合突变,c.-32-13T>G突变基因来源于母亲。B. Chr17:78084641 GAA基因测序显示,患者杂合变异,患者父亲杂合变异,患者母亲无变异,c.1551+2T>G突变基因来源于父亲。Fig.3 Chr17:78078341 and Chr17:78084641 GAA gene sequencing Maps
2 讨论
庞贝病是一种罕见的常染色体隐性遗传病,其特征是酸性α-葡萄糖苷酶水平降低或缺失造成溶酶体降解糖原受损[3]。庞贝病有两种主要分型:早发型庞贝病(infantile-onset Pompe disease,IOPD)没有残留酶活性,常于婴儿期发病;晚发型庞贝病(late-onset Pompe disease,LOPD)存留一定的酶活性,一般发病在儿童期、少年期或青年期。
晚发型庞贝病临床表型较多,即使在类似的遗传背景下也会有不同,如心脏扩大、呼吸功能不全、近端肌肉无力等,这可能是因为存在营养、环境等非遗传因素[4]。临床表型最常见累及下肢近端肌肉和轴肌,然后是上肢和呼吸肌[5]。实验室检查约95%患者晚发型庞贝病肌酸激酶升高,血清肌酸激酶范围通常从正常到正常上限的5倍[5]。电生理检查常规神经传导通常正常,针极肌电图70%表现为肌病模式[6]。定量肌肉核磁共振技术能够提供诊治价值[7],本例患者肢体肌肉磁共振提示肌炎,为进一步明确遗传性肌病提供了诊断思路。空泡性肌病是庞贝病经典病理改变。不过,即使肌肉活检结果正常也不能完全排除晚发型庞贝病,具有适当表型的患者仍需高度怀疑,因为最终诊断需要检测酸性α-葡萄糖苷酶,如果酶活性低于参考范围,可以进一步进行DNA分析,检测到GAA基因的两个突变可证实庞贝病的诊断[8]。本例患者GAA基因测序发现两个临床表型与庞贝病高度相关且致病性证据较为充分的基因变异,c.-32-13T>G和c.1551+2T>G分别来自母亲和父亲,上述两个致病变异最终造成基因功能丧失。
文献表明,已有600多个与庞贝病相关的遗传变异存在于“庞贝病GAA变异数据库[9]”。虽然该数据库允许在识别两个等位基因的致病变异之后预测患者的表型,但类似本例患者的两个等位基因致病变异及预测表型尚未见报道。因此,本比例可积累更多庞贝病患者的致病变异基因以不断完善数据库,促进研究发展。
参考文献:
1. 赵辰, 谢小雷, 冀维真. 美国二代测序技术临床应用的共识声明、实践资源、技术标准和指南的概述[J]. 中华医学遗传学杂志, 2021, 38(6): 513-520.
2. LEE D H, QIU W J, LEE J, et al. Hypertrophic cardiomyopathy in Pompe disease is not limited to the classic infantile-onset phenotype[J]. JIMD Rep, 2014, 17: 71-75.
3. REYES-LEIVA D, ALONSO-PéREZ J, MAYOS M, et al. Correlation between respiratory accessory muscles and diaphragm pillars MRI and pulmonary function test in late-onset Pompe disease patients[J]. Front Neurol, 2021, 12: 621257.
4. AUSEMS M G, TEN BERG K, BEEMER F A, et al. Phenotypic expression of late-onset glycogen storage disease type Ⅱ: Identification of asymptomatic adults through family studies and review of reported families[J]. Neuromuscul Disord, 2000, 10(7): 467-471.
5. TOSCANO A, RODOLICO C, MUSUMECI O. Multisystem late onset Pompe disease (LOPD): An update on clinical aspects[J]. Ann Transl Med, 2019, 7(13): 284.
6. AL-HASHEL J, ISMAIL I. Late-onset Pompe disease presenting with isolated tongue involvement[J]. Case Rep Neurol, 2022, 14(1): 98-103.
7. FIGUEROA-BONAPARTE S, LLAUGER J, SEGOVIA S, et al. Quantitative muscle MRI to follow up late onset Pompe patients: A prospective study[J]. Sci Rep, 2018, 8(1): 10898.
8. LLERENA JUNIOR J C, NASCIMENTO O J, OLIVEIRA A S, et al. Guidelines for the diagnosis, treatment and clinical monitoring of patients with juvenile and adult Pompe disease[J]. Arq Neuropsiquiatr, 2016, 74(2): 166-176.
9. DE FARIA D O S, ’T GROEN S L M I, HOOGEVEEN-WESTERVELD M, et al. Update of the Pompe variant database for the prediction of clinical phenotypes: Novel disease-associated variants, common sequence variants, and results from newborn screening[J]. Hum Mutat, 2021, 42(2): 119-134.
【引用格式】黄丽,张娇贵,陈定邦 ,等. 糖原贮积病Ⅱ型1例[J]. 中国神经精神疾病杂志,2024,50(10):602-604.
【Cite this article】HUANG L,ZAHNG J G,CHEN D B,et al.A case of glycogen accumulation disease typeⅡ[J]. Chin J Nervous Mental Dis,2024,50(10):602-604.
DOI:10.3969/j.issn.1002-0152.2024.010.006

