Nat Methods:结合微阵列和NGS的可扩展空间转录组学方法Array-seq,突破空间分析界限
时间:2024-12-22 12:01:45 热度:37.1℃ 作者:网络
研究组织中细胞和分子的空间组织结构是生物医学和临床病理学的一个基础性课题。近年来,空间转录组学(ST)的进展使得在组织切片特定区域进行转录组测序成为可能,并且出现了商业化平台,例如Visium。然而,现有的ST方法面仍存在可用表面积小、与苏木精和伊红(H&E)染色兼容性差、低通量和高成本等局限性,阻碍了其在基础和临床研究中被广泛使用。
近日,美国芝加哥大学的研究团队在Nature Methods发表了题为“Repurposing large-format microarrays for scalable spatial transcriptomics”的文章,报道了新开发的空间转录组方法Array-seq,其能够将经典的寡核苷酸微阵列重新应用到空间转录组学分析。研究团队利用携带定制探针的微阵列生成Array-seq载玻片,这些探针包含已知空间坐标上独特条形码的共同序列,并在微阵列上的所有点产生mRNA捕获探针。基于小鼠和人体组织切片,研究人员证明了Array-seq能够基于较大表面积的载玻片产生具有高检测灵敏度和定位特异性的高通量空间转录组信息。总之,通过结合经典的DNA微阵列和NGS技术,该团队创建了一个简单灵活的平台,可用于从小到大样本的大规模空间分子生物学研究。

文章发表在Nature Methods
主要研究内容
01 在定制微阵列上组装mRNA捕获探针
首先,研究人员设计了定制的寡核苷酸微阵列,其空间条形码序列两侧是常用序列,可以在载玻片上组装用于ST分析所需的mRNA捕获探针,定制微阵列是用3'锚定的寡核苷酸合成的,并且每个位点携带特有的空间条形码。此外,两侧有两个序列,分别为锚点1和锚点2,它们在微阵列上所有位点上都是常见的。
锚点1的序列通过其3'端连接到载玻片上,与测序引物的前24个碱基匹配,并作为cDNA扩增和Illumina测序的引物;锚点2的序列与常用的M13 forward(M13F)引物匹配。对于空间条形码,研究人员设计了974016个独特的序列,同时最大限度地减少了自二聚化和与两个锚点序列的相互作用。
接下来,为了在微阵列上组装ST探针,研究人员首先将两个寡核苷酸阵列杂交,其次使用DNA聚合酶在杂交锚点1的3'端合成空间条形码的反向补体,并使用DNA连接酶将新合成的条形码连接到锚点2的磷酸化5'端,利用DNA聚合酶Phusion优化间隙填充。为了提高检测敏感性,还清除了未连接的锚点,最终得到纯度大于75%的mRNA捕获探针。
由此产生的ST兼容阵列携带974016个直径为30µm的点,分布在1068行和912列,总面积为11.31cm2,可用于ST分析,阵列上的每个点在已知的x和y坐标上携带一个唯一的空间条形码序列用于定位,UMIs用于定量,寡核苷酸用于捕获聚腺苷化的转录本。
为了评估Array-seq切片上每个点捕获的细胞数量,研究团队计算了十种小鼠器官类型中与每个点重叠的组织切片中的细胞核数量,其中肌组织和脑组织的中位细胞核数为2和3,骨髓和脾脏的中位细胞核数为15和19,其他组织类型,包括棕色脂肪、肝脏、心脏、肺、结肠和肾脏,每个斑点显示的细胞核中位数在5到8之间。因此,一张Array-seq载玻片可以捕获200万-2000万个细胞,这主要取决于组织类型和要分析的组织切片的数量和大小。
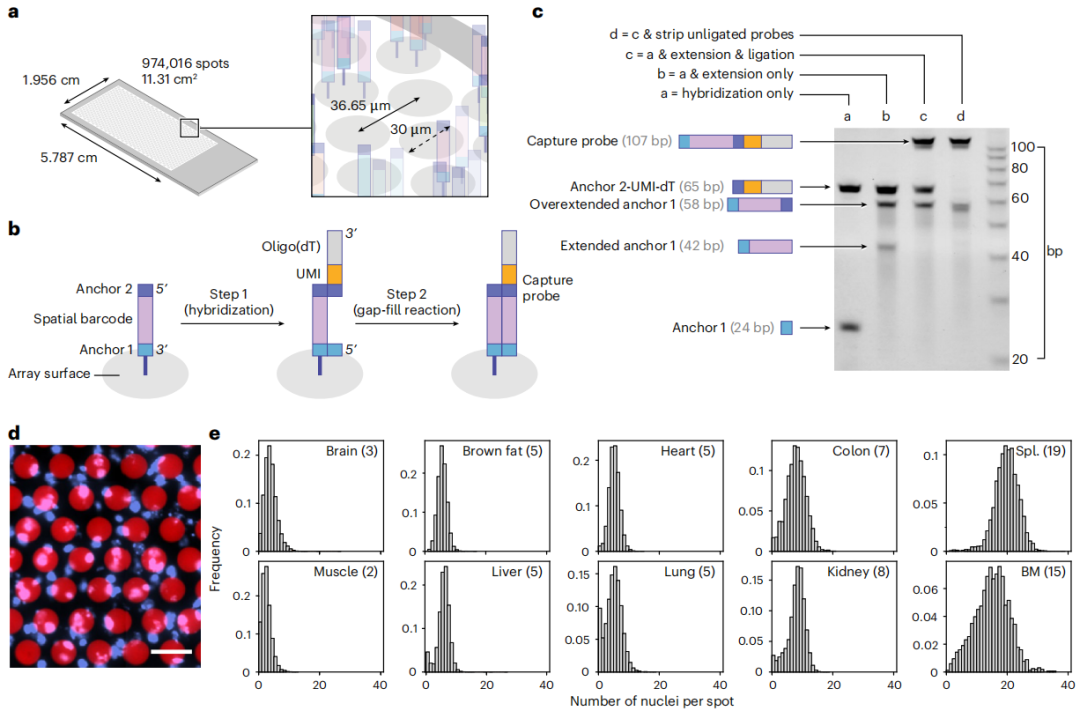
图1. 利用微阵列在载玻片上组装空间条形码mRNA捕获探针
02 Array-seq产生高质量的ST数据
为了测试Array-seq,研究团队首先分析了小鼠大脑的主嗅球(MOB)系统,由于其尺寸较小且具有明确的形态分层,常常作为ST方法基准测试时选择的组织。研究在阵列上检测到平均3582.2个UMIs和1971.5个基因。接下来,使用Leiden算法对MOB数据进行无监督聚类分析,该算法识别出与MOB组织所有已知层对应的聚类。此外,MOB组织层标记基因的空间分布与前期已经发表的原位杂交数据也保持一致,还与H&E成像数据定义的组织区域一致。
此外,研究还检测了不同细胞类型在MOB组织中的空间分布,发现在斑点上标注的细胞类型被正确地分配到特定的MOB层。以上数据表明,Array-seq能够生成高质量ST谱,并以高分辨率检测基因、细胞类型和组织学区域。同时,比较分析发现Array-seq在斑点密度、直径和总表面积等指标上优于ST分析的金标准平台Visium,同时保持了相似的灵敏度和特异性。
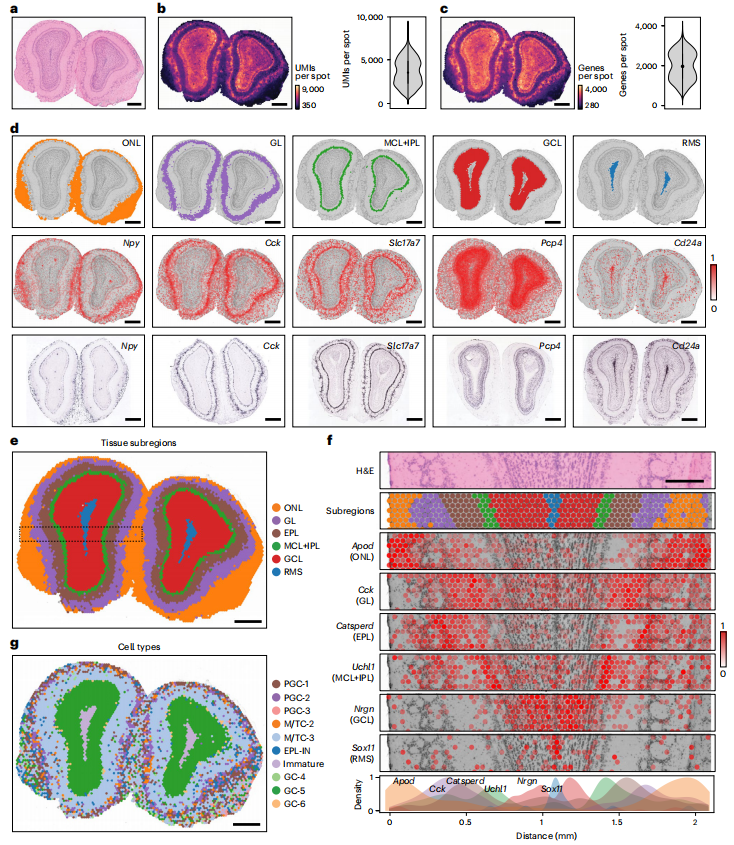
图2. Array-seq准确捕获组织中区域特异性表达模式
03 Array-seq可实现三维层面的高通量分析
研究团队使用小鼠肾脏作为测试系统,检测了Array-seq用于空间转录组三维分析的性能。Array-seq捕获区的表面积允许在同一载玻片上平行分析一系列小鼠肾脏切片,为了验证这一点,研究人员通过对小鼠肾脏进行连续切片,在总计约800μm的组织深度上每80-120μm分析一个切片,生成了8个10μm厚的切片。通过对相同组织切片上的Array-seq数据进行无监督聚类分析,H&E观察到的肾脏三维组织学特征得到了正确的复原,每个组织亚区覆盖的表面积根据肾内给定切片的深度而变化。
此外,通过无监督聚类分析并经组织学证实的每个组织亚区标记基因的空间分布与其预期的区域表达也密切相关。因此,结果表明,Array-seq载玻片的大表面积能够以高分辨率和高通量生成空间转录组学数据,包括三维空间转录组分析。
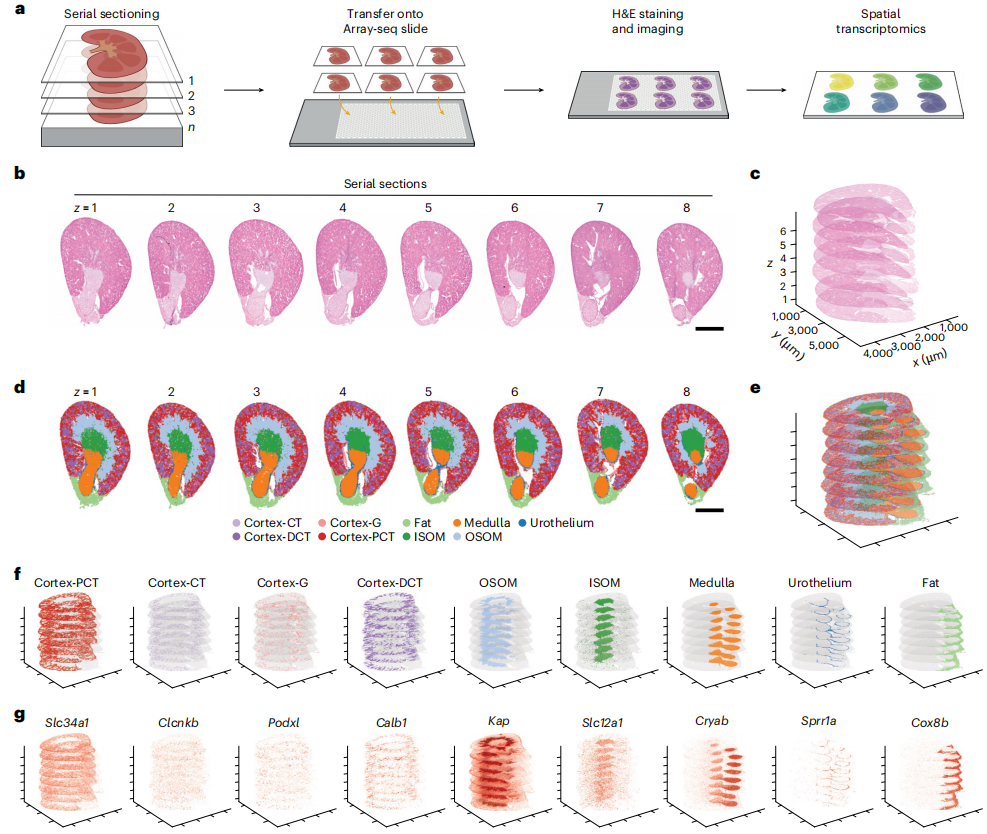
图3. Array-seq可实现空间转录组的三维分析
04 Array-seq可实现人类器官全视野切片分析
理论上,Array-seq的大表面积可以用于分析来自较大组织的整片切片。为了验证这一点,研究团队获得了新鲜的人脾组织,并对其进行了Array-seq分析。H&E染色和人体脾脏切片成像显示了已知的脾脏组织学结构。对与H&E分析的同一脾脏切片获得的ST数据进行聚类,发现Array-seq能正确识别出红髓、白髓和边缘区。随后,研究分析了趋化因子对及其匹配受体的空间表达模式,发现编码趋化因子和趋化因子受体的基因空间分布与其在脾组织中的预期定位一致。
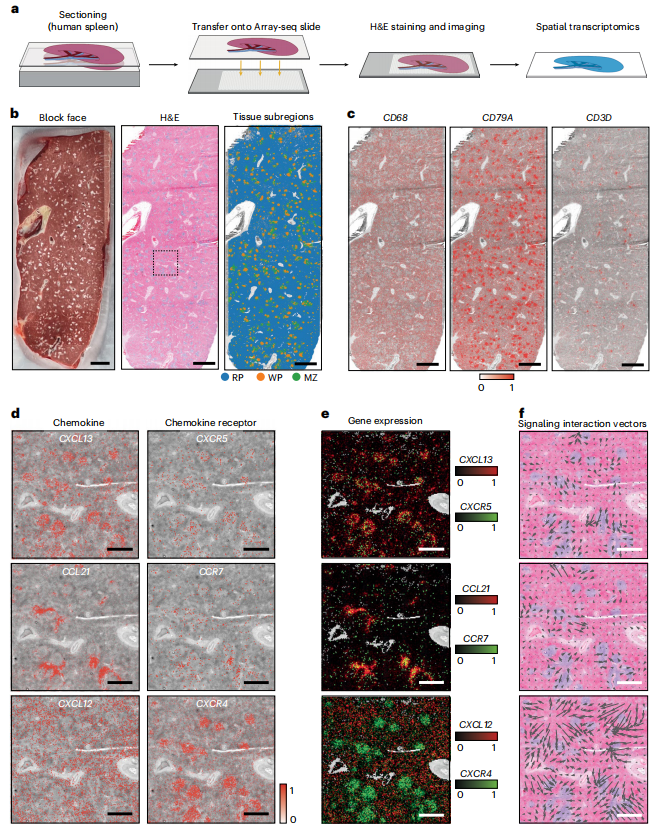
图4. 人类脾脏整个切片的Array-seq分析
结 语
综上所述,该研究将微阵列与NGS技术相结合,开发了一种简单有效的空间转录组学分析方法Array-seq,其能够突破空间分析界限,处理从小到大的样本,例如,从小鼠组织到整个人体器官等。同时,Array-seq还可以通过处理来自同一组织的大量连续切片来实现组织样本的三维分析。值得注意的是,Array-seq具有较大表面积、成本低且具有可扩展性,无需特殊专业知识或仪器即可采用,并且与H&E染色兼容,因此可适用于所有基础和临床研究领域。在未来,Array-seq平台将通过实现大规模的空间分子学研究为空间组学革命做出贡献。
论文原文:
Cipurko, D., Ueda, T., Mei, L. et al. Repurposing large-format microarrays for scalable spatial transcriptomics. Nat Methods (2024). https://www.nature.com/articles/s41592-024-02501-5

