病例分享 | SMARCB1/INI-1缺失的差分化脊索瘤一例
时间:2024-12-19 12:01:14 热度:37.1℃ 作者:网络
01 病史介绍
患者女,18岁,以“间断性腰痛半年,加重1月”为主诉。半年前无明显诱因出现间断性腰痛,无双下肢放射痛,症状时轻时重;1月前因劳累后感腰痛症状加重,伴双下肢麻木无力,自服止痛药物治疗效果差。当地医院影像学示L1、L4、L5、骶骨、髂骨及周围软组织病变,考虑嗜酸性肉芽肿?,为了解肿物性质来我院就诊。既往体健。
02 专科检查
右腋后、右乳腺外上象限、右胫骨近端外侧均可触及一肿物,大小不等,较大者约3x2.5cm,隆起于表皮;骶尾部可触及一肿物,直径约10cm,边界不清,稍隆起于皮肤,皮温正常,压痛(+)。各肌群肌力V级,肌张力正常。
03 辅助检查
PET-CT示:
1.骶尾骨巨大软组织肿块影,PDG代谢不均匀性活跃,局部虫噬样骨质破坏伴周围软组织形成,分界不清,病灶向外生长侵犯左侧臀中肌、臀大肌,向前突入盆腔,多考虑骨原发性肿瘤(脊索瘤?或其他?)侵及邻近肌肉可能。
2.L2椎体周围、L3椎体后方、L4椎体附件、L5椎体右不规则软组织影,代谢活跃,部分病灶侵入椎管内。
3.肋骨多发骨转移可能。
4.多部位淋巴结继发恶性肿瘤转移;后行左侧臀部肿物穿刺活检术。
04 大体所见
左侧臀部肿物穿刺活检术:
左臀部肿物:送检灰白灰红条索状组织10条,长0.4cm-1.2cm,直径0.1cm,全取。
05 镜下形态
组织内见呈成片或巢片状分布增生的上皮样细胞,细胞轻度到中度异型性,核圆形或椭圆形,部分核不规则、扭曲,核分裂像活跃(>5个/10HPF),可见片状、多灶坏死,部分肿瘤细胞胞浆可见空泡,局灶胞质嗜酸性,似横纹肌样细胞,缺乏脊索瘤常见的分叶状结构、“空泡状”细胞和黏液样基质。间质内可见散在的淋巴细胞、浆细胞等炎细胞。
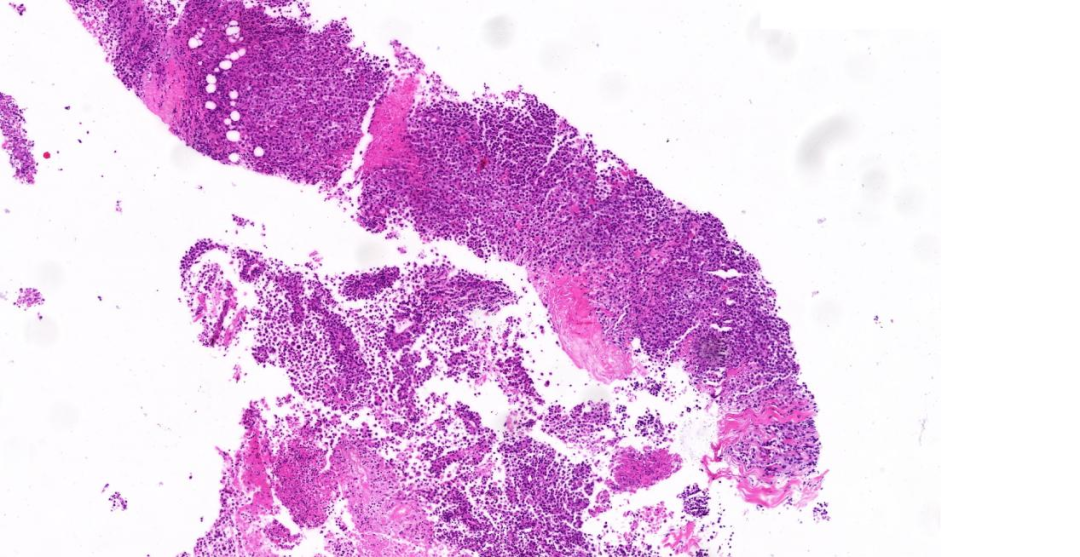
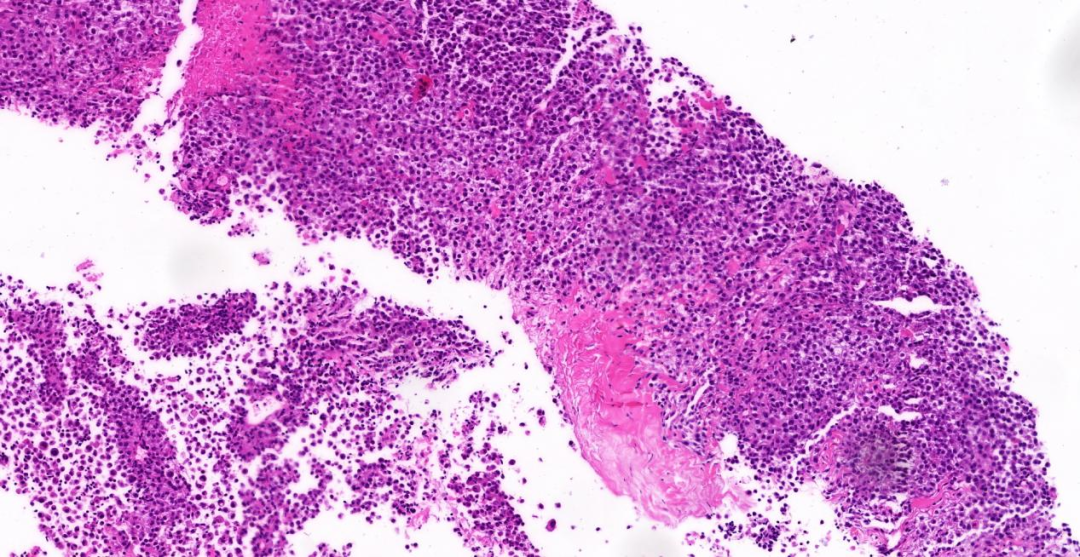
图1:低倍镜(5 x),上皮样的肿瘤细胞呈圆形或椭圆形弥漫性生长,局灶呈横纹肌样形态,伴周围片状坏死;
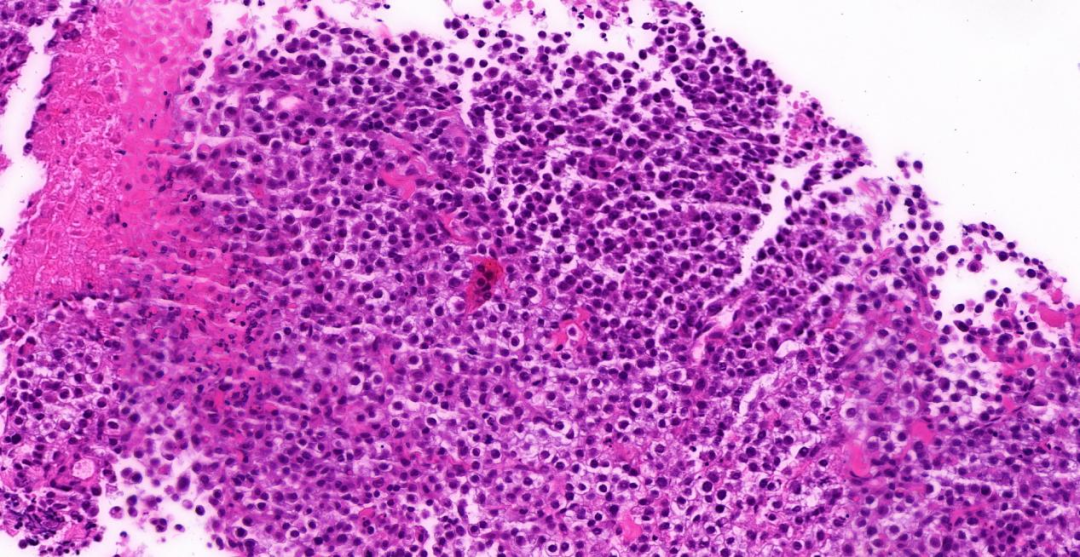
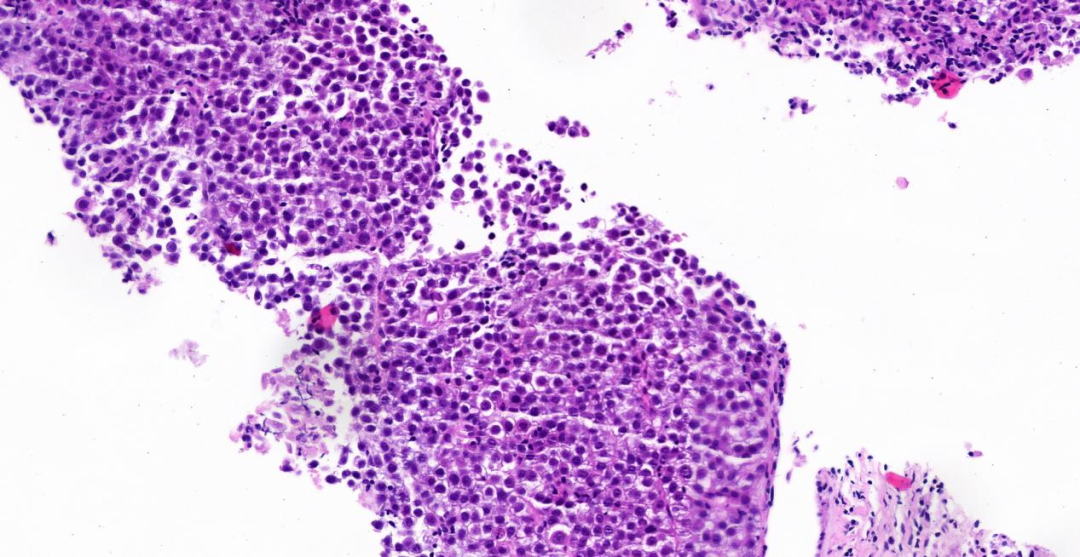
图2,3:中倍镜(20 x),肿瘤细胞胞浆透亮、少许空泡状,部分区域肿瘤细胞胞浆嗜酸性;少量淋巴细胞、浆细胞分布;
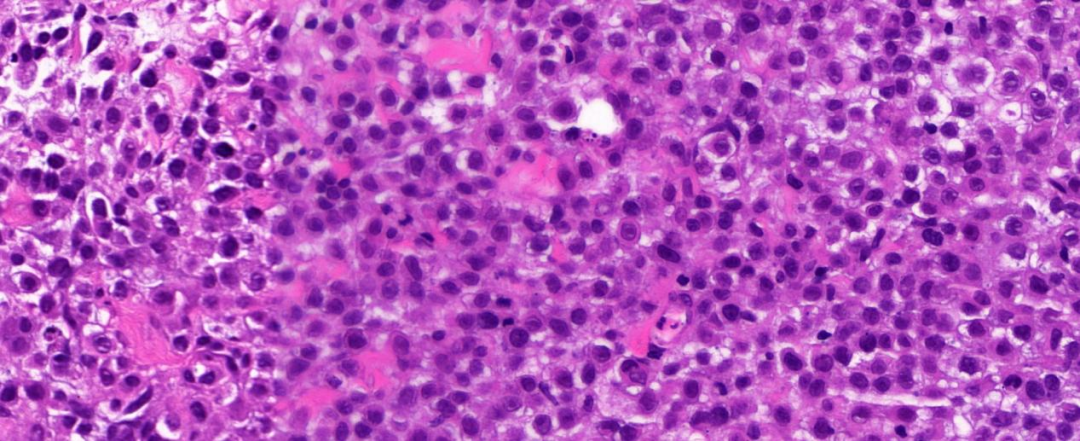
图4:高倍镜(40 x),肿瘤细胞部分细胞核偏位、核仁不规则,部分呈环状;局灶可见病理性核分裂;
06 免疫组化结果
肿瘤细胞弥漫强表达广谱CK(图5)、转录因子Brachyury(图6),局灶表达CD38和CD138,不表达Vimentin、SOX-10、S-100、CD34、Pax-8、Desmin、LCA、CD30、SALL4、CD1α、SMA、TTF-1、GATA-3,可见SMARCB1/INI-1核表达缺失(图7),肿瘤细胞Ki-67阳性增殖指数较高,约60%。
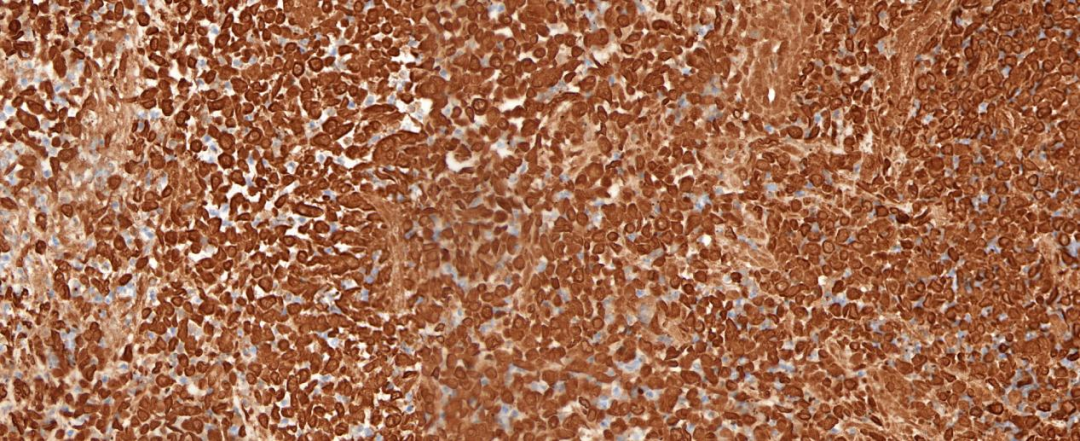
图5 肿瘤细胞弥漫表达CK;EnVision法 中倍放大;
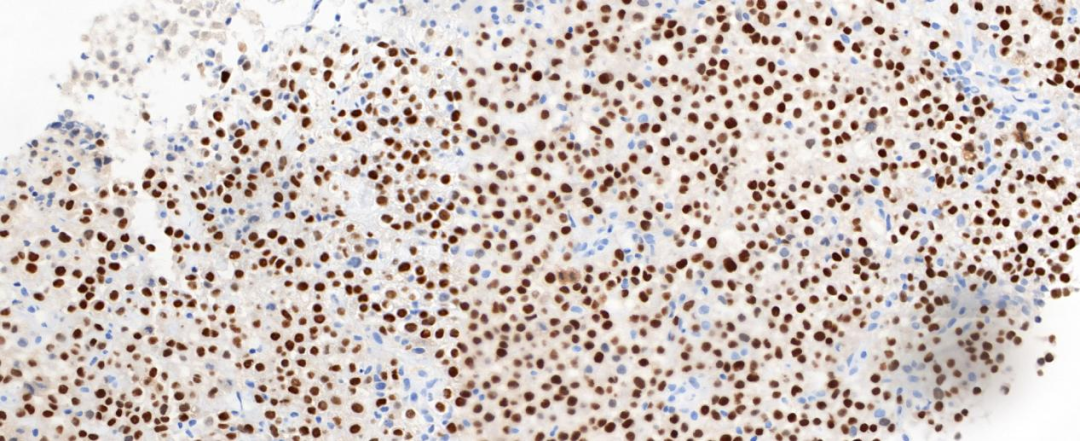
图6 肿瘤细胞核弥漫表达Brachyury;EnVision法 中倍放大;
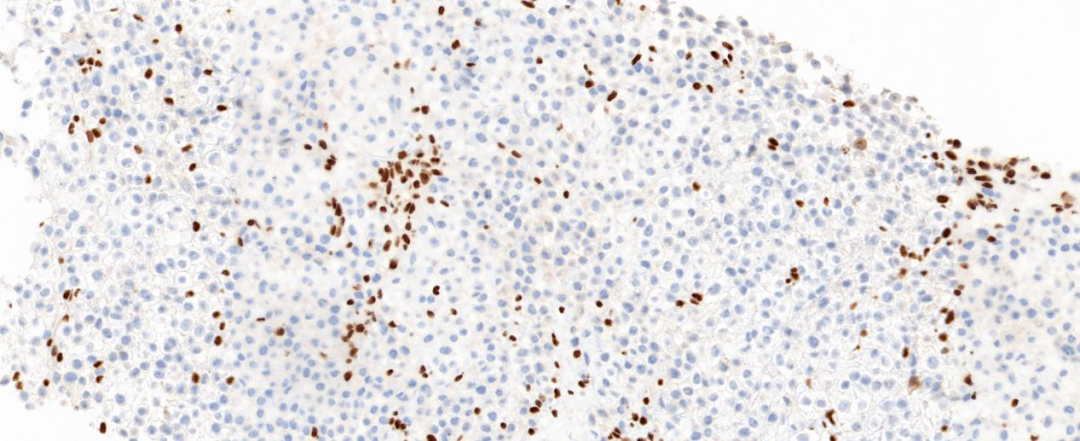
图7 肿瘤细胞INI-1蛋白核表达缺失;周围少量淋巴细胞及内皮细胞为阳性对照;EnVision法 中倍放大;
07 病理诊断
(左臀部肿物)SMARCB1/INI-1缺失的差分化脊索瘤(Poorly Differentiated Chordoma,PDC)。
讨 论
01 定义
定义 PDC是一种伴有脊索分化的差分化肿瘤,通常发生于中轴骨,特征是有SMARCB1的表达缺失。
ICD-O编码:PDC:9370/3
02 临床特征
脊索瘤(Chordoma)是临床少见的生长缓慢、侵袭性的骨肿瘤,主要发生于中轴骨,特别是颅底、脊椎、骶骨部位,起源上与胚胎脊索残余有关。脊索瘤最常见的临床症状是疼痛及部位周围神经病变。发病年龄上以成年人为主,罕见发生于儿童及年轻人。第五版WHO软组织和骨肿瘤分类中将脊索肿瘤分为4个类型:良性的脊索细胞瘤(Benign notochordal cell tumor)、经典型脊索瘤(Conventional chordoma)、去分化脊索瘤(Dedifferentiated chordoma)、差分化脊索瘤(Poorly Differentiated Chordoma,PDC),并认为PDC是具有SMARCB1/INI-1缺失的特殊类型脊索瘤。PDC主要发生于儿童与青少年,平均发病年龄9.7岁,女性患者大约是男性患者的2倍,以中颅底为常见的发病部位,尤其是斜坡,肿瘤平均直径4.3 cm(1. 5~12 cm)。目前,该病文献报道不足100例。
03 影像学特征
CT 扫描主要表现为边界尚清地图样骨质破坏,伴高密度或等密度软组织肿块,部分伴有钙化,发生于斜坡的病变可以延伸至颈椎;头颅MRI扫描,T1和T2加权像肿瘤呈不均匀结节性信号影,增强后呈中等或显著不均匀强化,部分明显强化伴蜂窝状低信号影。
04 病理学特征
1. 大体特征:
肿瘤大小直径从2cm到>10cm不等,大多数直径在5cm左右。切面呈灰红、灰白色,质中,局灶见出血及坏死,局部见骨组织,质硬。
2.镜下表现:
低倍镜下见肿瘤边界不清,侵袭性生长,被纤维组织增生分隔为小叶状,成片实性或巢团状分布增生的上皮样、横纹肌样细胞,部分呈梭形,缺乏脊索瘤常见的分叶状结构、较大的“空泡状”细胞和黏液样基质,个别病例形态类似经典脊索 瘤。高倍镜下肿瘤细胞轻度到中度异型性,核圆形或椭圆形,部分核不规则、扭曲,核分裂像活跃[可达12个/10HPF],可见片状、多灶坏死,少部分肿瘤细胞胞浆可见空泡,局灶胞质嗜酸性,似横纹肌样细胞;另外局部见卵圆形细胞区,呈漩涡状排列,似脑膜瘤形态;个别病例见幼稚骨组织化生,间质内可见散在的淋巴细胞、浆细胞等急慢性炎细胞浸润。
3.免疫表型:
PDC表达经典脊索瘤常见免疫标志物,如广谱蛋白CK、EMA、S-100蛋白及波形蛋白,以及脊索瘤特异性及灵敏的标志物Brachyury,伴有特征性的SMARCB1/INI1 核表达缺失;其中Brachyury对各型脊索肿瘤的诊断有高度特异度和灵敏度。P53蛋白可高表达,Ki-67阳性增殖指数范围10%-60%。通常不表达GFAP、Oligo-2、STAT-6、PR、SSTR2A、D2-40、Desmin。
05 分子遗传学
SMARCB1/INI1基因位于染色体 22q11. 2,是SWI/SNF复合物的主要核心亚基。在ATP依赖性染色质重塑中起作用,从而调控基因表达,参与多种恶性肿瘤发生,也被命名为SMARCB1/INI1缺失性肿瘤,如上皮样肉瘤、上皮样恶性外周神经鞘膜瘤、差分化脊索瘤、中枢神经系统非典型畸胎样/横纹肌样肿瘤(AT/RT)等。不同于AT/RT的SMARCB1/INI1基因位点上点突变,PDC则为由SMARCB1位点所在的22q纯合性或杂合性缺失导致的SMARCB1失活。一些研究显示,SMARCB1缺失是经典型脊索瘤早期发生事件,可以通过基因组学改变(如基因组倍增)进展为PDC。文献报道,PDC存在EWSR1基因的纯合/杂合性缺失,可与SMARCB1共缺失。DNA甲基化分析显示,PDC不同于经典型脊索瘤和AT/RT,而是具有独特的DNA甲基化谱系特征的独立肿瘤实体。其他的遗传学改变包括包括TBXT拷贝获得、单核苷酸变异(single nucleotide variation,SNV)等。
06 治疗与预后
PDC临床上主要是手术、放射、化疗相结合的方法治疗。相比成人发生的脊索瘤,PDC更具有侵袭性、预后差,并具有高病死率。由于报道的病例较少,需要更多的病例研究探寻有效的治疗策略。
07 鉴别诊断
需要与INI-1缺失性肿瘤及脊索瘤其他类型等鉴别,主要肿瘤如下:
(1)儿童AT/RT:与发生颅底的AT/RT相鉴别,AT/RT可见原始神经外胚层成份及横纹肌样成份相混合,通常表现为多向分化,上皮样成份相对较少。免疫组化上都存在SMARCB1/INI1的缺失,但Brachyury核阳性表达有助于二者鉴别。
(2)去分化脊索瘤:组织学上可见双相分化,由经典型脊索瘤和高级别梭形细胞肉瘤组成,肉瘤区域界限较清晰,转化突然;免疫组化二者也不同,因此组织形态学加免疫组化或分子检测可以鉴别二者。
(3)经典型脊索瘤:典型的分叶状结构,肿瘤细胞呈片状、条索状分布于黏液样基质中。常常见到“泪滴状”细胞形态,胞质空泡样、脂肪细胞样,免疫组化无SMARCB1/INI1的缺失。因此,组织学形态和免疫组化可将二者鉴别。
(4)上皮样肉瘤:组织学呈肉瘤样形态,表达上皮标记物,如广谱CK,EMA,及Vimentin,但Brachyury核阴性表达,可以鉴别。
(5)上皮样恶性外周神经鞘膜瘤:常见于成人的四肢近端和躯干,罕见于颅底,67%的病例存在SMARCB1/INI1 表达缺失,S-100蛋白多为弥漫性表达、不同程度的GFAP和 EMA阳性、广谱CK和 Brachyury核阴性表达可以帮助鉴别。
(6)儿童脑膜瘤:要与发生在颅底的脑膜瘤鉴别,二者在免疫组化和分子上不同。脑膜瘤常表达EMA、PR及SSTR2a而CKpan、Brachyury为阴性。分子上脑膜瘤可有P16缺失、TERT和NF基因突变,但无SMARCB1基因改变。
(7)其他骨或软组织的梭形细胞肿瘤:通常无经典型脊索瘤的“泪滴样”细胞和黏液软骨样间质等成份,一般不表达广谱CK、EMA、Brachyury,但分子检测常常伴有基因断裂和重排,可以鉴别二者。
*本文(包括图片)均为作者投稿, 仅供行业交流学习用,不作为医疗诊断依据。
参考文献
[1] 段泽君,姚坤,马忠,胡泽娟,向磊,齐雪岭.儿童颅底SMARCB1/INI1缺失性差分化脊索瘤5例临床病理学观察[J].中华病理学杂志,2022,51(1):33-38.
[2] 崔雪娥,王丽君,管雯斌,储彩婷,包磊,汪登斌.儿童差分化脊索瘤的影像和病理特征[J].中华放射学杂志,2023,57(5):553-555.
[3] 邵立伟,孙爱群,刘鹏,宋欣,王辅林,石怀银.SMARCB1/INI1缺陷型差分化脊索瘤4例临床病理特征并文献复习[J].临床与实验病理学杂志,2021,37(5):565-569.
[4] WHO Classification of Tumours Editorial Board. WHO classification of tumours of soft tissue and bone[M]. 5th ed.Lyon, France: IARC Press; 2020.
[5] Wilson B G,Roberts C W.SWI/SNF nucleosome remodellers and cancer[J].Nat Rev Cancer,2011,11(7):481-492.
[6] Kohashi K,Oda Y.Oncogenic roles of SMARCB1/INI1 and its deficient neoplasms[J.Cancer Sci,2017,108(4):547-552.
[7] Curcio C, Cimera R, Aryeequaye R, et al. Poorly differentiated chordoma with whole-genome doubling evolving from a SMARCB1-deficient conventional chordoma: A case report. Genes Chromosomes Cancer. 2021 Jan;60(1):43-48.
[8] 黄瑾,杨婷婷,蒋智铭,等.脊索肿瘤48例的临床病理学分析[J]. 中华病理学杂志,2021,50(03):201-206.

