Circ Res:南开大学陈佺等发现内皮细胞FUNDC1缺乏导致肺动脉高压!
时间:2024-12-15 06:00:43 热度:37.1℃ 作者:网络
肺动脉高压(PH)与内皮功能障碍有关。然而,内皮功能障碍的原因及其对PH的影响仍不完全清楚。
2024年11月28日,南开大学陈佺、Liao Xudong、Lu Chengzhi共同通讯在Circulation Research在线发表题为“Endothelial FUNDC1 Deficiency Drives Pulmonary Hypertension”的研究论文,该研究发现内皮细胞FUNDC1缺乏导致肺动脉高压。
临床研究对象和动物模型肺血管中PH值降低的FUNDC1蛋白水平。Global Fundc1缺乏加重了PH,而其过表达具有保护作用。FUNDC1的作用是由内皮细胞而非平滑肌细胞介导的。此外,在出生后小鼠中诱导内皮性Fundc1的丢失足以自发地引起PH,而内皮性Fundc1的增加在发病前后都可以防止PH的发生。从机制上讲,Fundc1缺乏损害了内皮细胞的基础线粒体自噬,导致线粒体功能失调的积累,代谢重编程,向有氧糖酵解,假性缺氧和衰老,可能通过mtROS-HIF2α信号通路。随后,缺乏Fundc1的内皮细胞增加了IGFBP2(胰岛素样生长因子结合蛋白2)的分泌,从而驱动肺动脉重塑,从而刺激PH。最后,体内原理验证研究表明,通过靶向内皮细胞有丝分裂、假性缺氧、衰老或IGFBP2,可以显著改善PH。

人们一直致力于了解肺动脉平滑肌细胞(PASMCs)在PH发病中的作用,因为这些细胞构成了PH期间发生剧烈重塑的大部分血管壁。然而,新出现的证据暗示了血管内皮细胞(ECs)的关键作用。考虑到内皮在调节血管张力中的重要作用,内皮功能障碍有助于PH早期的血管收缩是合理的。丛状病变是晚期PH的一个标志,由持续增殖的ECs组成,突出了失调的ECs在PH发展和进展中的作用。然而,内皮功能障碍是PH的原因还是结果仍有争议。此外,目前尚不清楚是什么触发了PH中的内皮功能障碍,以及这与PASMCs异常增殖的关系。
线粒体在ATP合成、生物分子合成、信号转导和程序性细胞死亡中发挥重要作用。线粒体利用氧气通过氧化磷酸化(OXPHOS)过程燃烧代谢底物以产生ATP。在OXPHOS过程中,活性氧(ROS)是不可避免的副产品,功能失调的线粒体产生过多的ROS是有害的。为了维持线粒体稳态,细胞进化出一种复杂的机制,通过线粒体自噬去除受损或不需要的线粒体。值得注意的是,由于氧化损伤对所有线粒体都是不可避免的,因此需要持续的低水平线粒体自噬通量,称为基础线粒体自噬,以保持线粒体稳态。这种基础线粒体自噬对于不能通过细胞分裂稀释或分裂功能失调的线粒体的长寿细胞尤其重要。
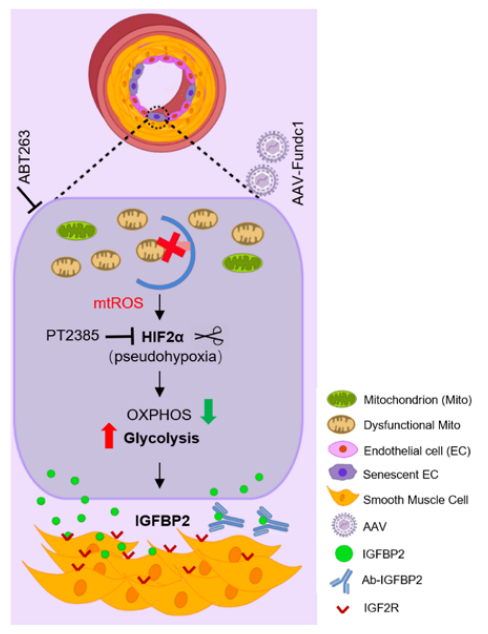
机理模式图(图源自Circulation Research)
线粒体功能障碍参与PH的发病机制。此外,慢性缺氧与PH密切相关,是肺功能障碍的主要原因或次要反应。已有研究已经确定线粒体外膜蛋白FUNDC1是一个关键的线粒体自噬受体,可被缺氧胁迫激活。在该研究中,研究人员证明了FUNDC1介导的基础线粒体自噬是内皮稳态的关键调节因子,其破坏导致代谢重编程和PH发病。与非PH对照组相比,小肺动脉中FUNDC1蛋白水平显著降低,这表明FUNDC1在PH中的潜在作用。
内皮细胞FUNDC1是PH发病机制的关键调节因子。出生后内皮细胞FUNDC1的缺失自发地刺激PH,而其过表达具有保护作用。相比之下,平滑肌FUNDC1是可有可无的。从机制上讲,内皮细胞FUNDC1的缺失会破坏线粒体自噬,并通过mtROS稳定HIF2α,导致代谢重编程、衰老和IGFBP2分泌,从而驱动PH。总的来说,该研究发现Fundc1介导的基底有丝分裂对内皮稳态至关重要,其破坏会引发PH发病。鉴于在PH患者中观察到类似的FUNDC1和IGFBP2变化,研究结果具有重要的临床相关性,并为PH提供了新的治疗策略。
参考消息:
https://www.ahajournals.org/doi/10.1161/CIRCRESAHA.124.325156

