【BJH】综述:慢性嗜酸性粒细胞白血病-非特指型
时间:2024-12-09 08:00:52 热度:37.1℃ 作者:网络
慢性嗜酸性粒细胞白血病-非特指型(Chronic eosinophilic leukaemia—not otherwise specified,CEL-NOS)是一种罕见的克隆性干细胞疾病,属于骨髓增殖性肿瘤(MPN),其特征为持续的嗜酸性粒细胞增多(HE)、预后不良以及进展为急性白血病的风险显著增加。对克隆性嗜酸性疾病(ED)的首次描述可追溯至 1912 年,当时Stillman发现了因恶性嗜酸性疾病导致严重外周血嗜酸性粒细胞增多的患者,并创造了术语“嗜酸性白血病”。在 20 世纪下半叶,通过纳入持久性HE病例,高嗜酸性粒细胞综合征(HES)的概念进一步加深了对ED的理解,定义为嗜酸性粒细胞计数>1.5×109/L,伴有嗜酸性粒细胞脱颗粒引起的相关器官损伤。
在过去十年中,在识别新的生物标志物和理解致病机制方面的重大进展,导致对DE的分类出现了几种新的分类,并对CEL-NOS的定义进行了完善。2021 年,国际嗜酸性粒细胞增多症合作工作组(ICOG-EO)公布了针对ED及其综合征的一系列共识定义(表 1)。ED目前根据临床特征和潜在病因进行分类,包括家族性嗜酸性粒细胞增多症(HEF)、意义不明的嗜酸性粒细胞增多症(HEUS)、继发性或反应性嗜酸性粒细胞增多症(HER)以及原发性嗜酸性粒细胞增多症(HEN)。应根据2022年世界卫生组织(WHO)分类或2022年国际共识分类(ICC)的最新更新来定义HEN的病因。

分子诊断的改进增强了对ED的分类,可以更精确地识别克隆性,并描绘出其他具有遗传学差异的亚型,例如伴有嗜酸性粒细胞增多和酪氨酸激酶基因融合的髓系/淋系肿瘤(myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase gene fusions,MLN-TK)。因此,更新的WHO分类将“NOS”(非特指型)这一名称从 CEL 中移除,而ICC则保留了这一名称,因为 MLN-TK 可能呈现出类似 CEL 的临床或形态学特征。本文始终使用后者。尽管如此,无论采用何种分类系统,CEL-NOS 仍是一种排除性诊断,其与特发性嗜酸性粒细胞增多症(iHES)的鉴别诊断是基于通过细胞遗传学和分子检测证明的克隆性,和/或在周围血或骨髓中存在过量原始细胞。
《British Journal of Haematology》近日发表综述,总结了关于 CEL-NOS 的现有知识,包括诊断检查、基因组进展以及该领域持续存在的治疗挑战;旨在提供关于 CEL-NOS 的广泛视角,并确定未来研究的关键领域。

发病机制
嗜酸性粒细胞生物学
嗜酸性粒细胞是白细胞的一个特殊亚群,参与多种免疫反应,包括过敏反应和寄生虫感染。嗜酸性粒细胞的名字源于对酸性染料的独特染色,该现象最早由Paul Ehrlich于1879年发现。从形态上看,嗜酸性粒细胞的特点为细胞核呈分节状,通常为两叶状,核与胞浆的比例通常为1:3。嗜酸性粒细胞通常占外周血白细胞的1%至6%,绝对嗜酸性粒细胞计数(AEC)通常介于0.05至0.5×109/L。
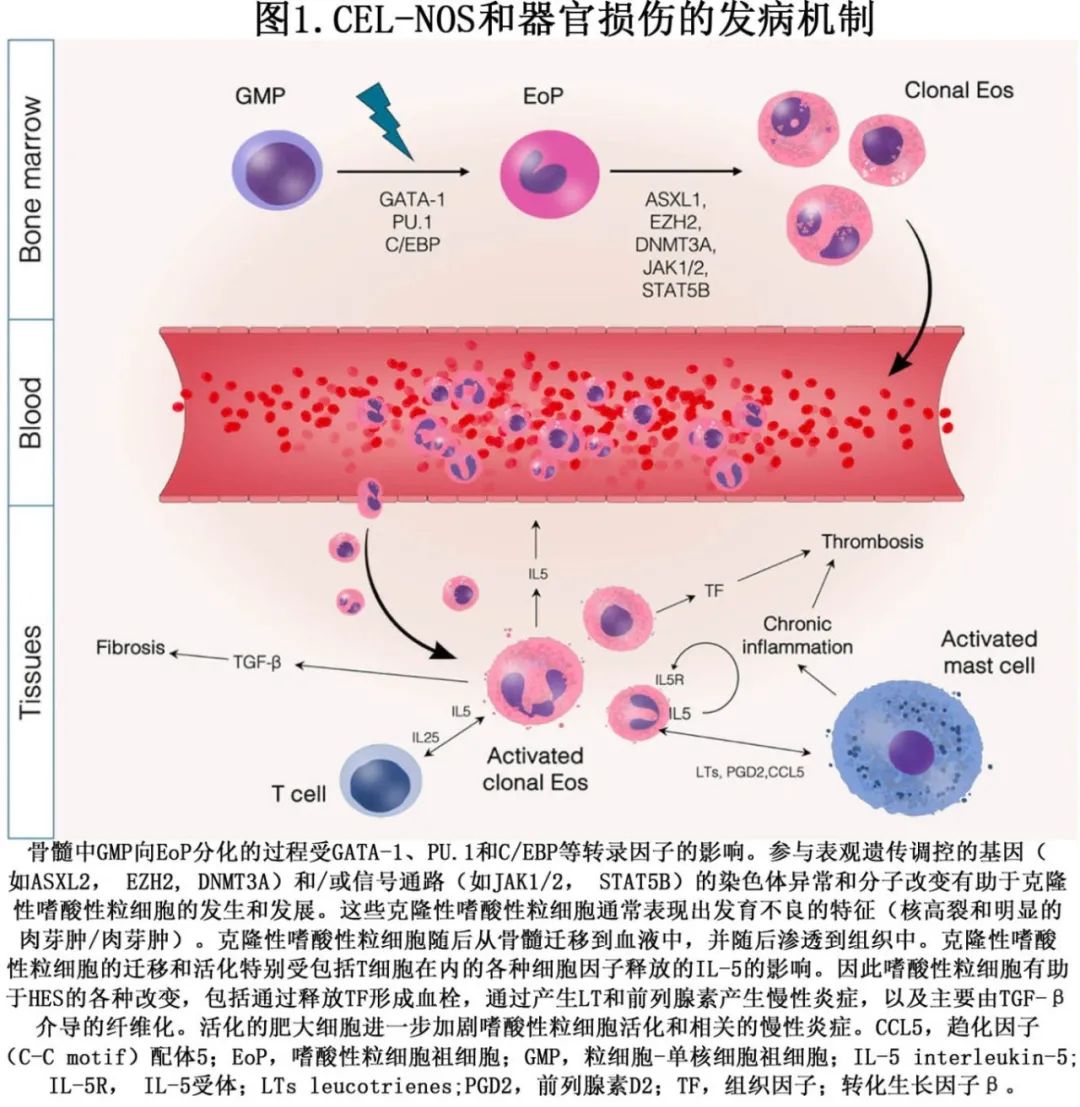
在人类中,嗜酸性粒细胞起源于共同的 CD34+髓系祖细胞(图1),其成熟和分化受到多种调节细胞命运和基因表达的因素的影响。参与该过程的关键转录因子包括 GATA结合蛋白-1(GATA-1)、PU.1和 CCAAT/增强子结合蛋白(C/EBP)。此外,粒细胞-巨噬细胞集落刺激因子(GM-CSF)、白细胞介素-3(IL-3)和白细胞介素-5(IL-5)等因子也协同参与成熟嗜酸性粒细胞的发育。
CEL-NOS中的基因突变和通路失调
CEL-NOS中的克隆性嗜酸性粒细胞增多是由于发生肿瘤性增殖,是干细胞起源髓系肿瘤的一部分(图1)。与其他经典骨髓增殖性肿瘤相比,CEL-NOS缺乏特定的细胞遗传学或分子学异常,然而CEL-NOS中嗜酸性粒细胞的不受控制的克隆性增殖似乎与基因突变有关,这些基因突变破坏了正常的细胞生长和分化机制。
分子谱分析经常揭示参与关键调节通路的基因突变,例如与表观遗传调节相关的基因(如 ASXL1、DNMT3A、EZH2 和 TET2)的突变,以及与剪接体相关的基因(如 SRSF2 和 SF3B1)的突变,经常见于报道。此外,已有五例携带 KITM541L 突变的CEL-NOS报道,显示对低剂量伊马替尼治疗敏感。JAK/STAT 通路(对嗜酸性粒细胞生成和存活至关重要)的突变也有报道。这些突变可能独立于其他基因改变而驱动原发性嗜酸性粒细胞增多症。在伴有嗜酸性粒细胞增多症的髓系肿瘤以及多达 15%的CEL-NOS患者中,已报道 STAT5B 基因的激活突变,特别是 N642H 突变。此外,在同时符合CEL-NOS和真性红细胞增多症(PV)诊断标准的患者中,已描述了JAK2e13InDel 突变;该突变会导致 JAK2 的组成型激活,即使在缺乏其配体的情况下,也能增强 IL-3、IL-5 和 GM-CSF 受体的信号传导。2021 年,在一例CEL-NOS病例中发现了一种新型激活 JAK1 的突变(R629_S632delinsSA)。该突变可激活 JAK/STAT 通路,导致不依赖于 IL-3 的嗜酸性粒细胞增殖,并且对 JAK 抑制剂芦可替尼敏感。
HES
在CEL-NOS中,不受控制的克隆性增殖导致外周血和组织中的嗜酸性粒细胞显著增加,从而引发各种临床并发症(图 1)。嗜酸性粒细胞功能的复杂性及其与其他炎症细胞的相互作用,连同细胞因子的释放,导致HE的临床病程差异极大。嗜酸性粒细胞可浸润并损害多种组织,包括皮肤、肺、胃肠道、心脏和大脑。这一过程由多种表面受体介导,这些受体对于黏附、归巢和组织迁移至关重要。响应于IL-5,嗜酸性粒细胞被动员进入外周循环,并通过趋化分子(如C-C基序配体11[CCL11])、CCL24 和 CCL26)与其表面受体(包括 CCR1 和 CCR3)的相互作用被引导至目标组织。
受到IL-4、IL-5和IL-13的刺激后,嗜酸性粒细胞会释放多种颗粒蛋白,包括主要碱性蛋白(MBP)1 和 2、嗜酸性粒细胞阳离子蛋白(ECP)、嗜酸性粒细胞过氧化物酶(EPX)、嗜酸性粒细胞衍生神经毒素(EDN)以及Charcot–Leyden晶体蛋白/半乳糖凝集素-10(CLC/Gal-10)。这些分泌的蛋白质,连同其他趋化因子、细胞因子和介质,会驱动白细胞募集、炎症和纤维化。在这种情况下,转化生长因子β(TGF-β)在组织重塑和纤维化中起着关键作用。此外,嗜酸性粒细胞可释放胞内 DNA 以形成嵌入颗粒蛋白的网状细胞外诱捕物(extracellular traps,ET)。ET 连同释放的有毒蛋白质,会影响内皮细胞,并促进血小板活化和聚集,可能导致血栓形成。
疾病概述
流行病学
由于其罕见性和异质性临床表现,关于CEL-NOS基于人群的数据有限,导致其流行病学特征定义不清。在美国,报告的年龄校正发病率为0.33/ 10万人/年,在年龄≥60 岁的个体中则上升至0.087/10万人/年。诊断时的中位年龄为50-60 岁,男性和白人多于非洲裔美国人或其他种族。随着年龄的增长发病率增加可能是由于基因和表观遗传突变逐渐累积导致克隆性嗜酸性粒细胞扩增,而激素或遗传因素可能导致性别差异。
临床表现
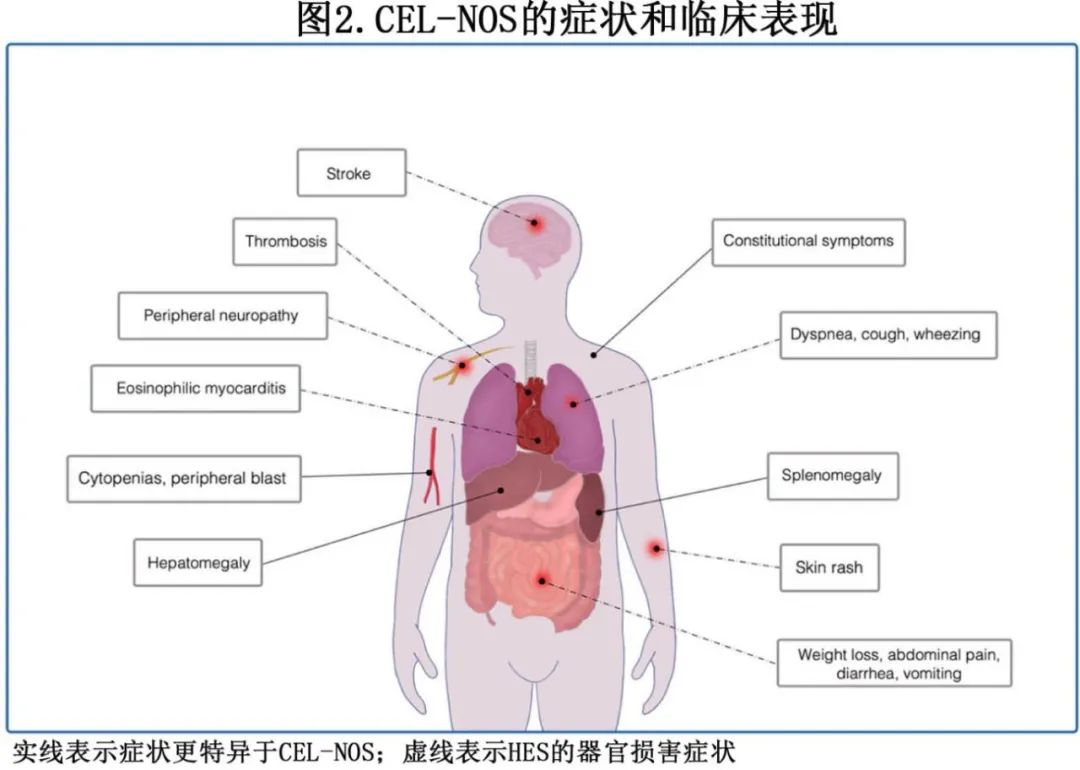
除了HEUS(其定义为无症状性)外,HE的临床表现与基础疾病并不严格相关。与嗜酸性粒细胞增多相关的症状可能与其他疾病相似,导致仅基于临床表现而进行诊断较为复杂。此外, CEL-NOS的临床表现差异很大,并受到嗜酸性粒细胞增多程度和器官受累情况的影响。从历史上看,CEL是一组以血液学症状为主的HES患者,症状包括血细胞减少、脾肿大、血液和骨髓中未成熟的嗜酸性粒细胞前体细胞以及原始粒细胞增多(图 2)。在一项纳入 125 例HE患者的队列研究中,对来自一家三级肿瘤学中心的 21 例CEL-NOS患者进行了症状分析,包括全身症状(38%)、皮疹(33%)、脾肿大(24%)以及HE相关的其他器官病变(52%)。其他证据报告称,70%的CEL-NOS患者有脾肿大,60%的患者有肝肿大。梅奥诊所17 例CEL-NOS患者中分别有 17%和 35%报告持续咳嗽和胃肠道症状(如腹泻、腹痛、恶心和呕吐)。此外高达 20%的CEL-NOS患者中有心脏受累的报告。然而这些表现也可能出现在其他形式的HE中,包括iHES。临床表现有显著重叠,也凸显了准确诊断ED的复杂性。
实验室结果
根据定义,CEL-NOS患者存在外周血HE(≥1.5×109/L),嗜酸性粒细胞占白细胞总数的比例>10%。白细胞计数介于15 至 35×109/L,但也有超白细胞增多症的报告。平均嗜酸性粒细胞计数介于4.1 至 22.1×109/L。可能会出现学细胞减少的情况,包括贫血和/或血小板减少,或者血小板增多。已报道的血清学异常包括乳酸脱氢酶(LDH)水平升高、免疫球蛋白 IgE 水平升高、维生素 B12 水平升高以及类胰蛋白酶水平升高。
血涂片通常显示成熟的嗜酸性粒细胞,具有非典型性,主要特征为核多分叶和明显的颗粒减少或缺失,伴有少量更不成熟的细胞。骨髓组织病理学通常显示骨髓增生异常综合征(MDS)、MDS/骨髓增殖性肿瘤(MPN)或 MPN 的典型变化。骨髓多为细胞增多,髓系与红系比例显著增加。嗜酸性粒细胞增多,有时不典型,同时伴有其他异常,如微巨核细胞(micromegakaryocytes)、不同程度的粒细胞生成异常(dysgranulopoiesis)和红系异常(dyserythropoiesis)以及中重度纤维化。
基因异常
细胞遗传学异常较常见,并且是克隆性的关键标志,常见的异常包括 8 号染色体三体、5 号、7 号和 12 号染色体的缺失或异常以及复杂核型。在细胞遗传学分析未能检测到异常的情况下,二代测序(NGS)可在超过 30%的原本归类为HES的病例中确定克隆性。NGS 的发展已经确定了常见于MPN的基因突变,如 ASXL1、TET2、EZH2 和 DNMT3A,以及 JAK/STAT 等信号通路。对 64 例非MLN-TKHESN异常的 NGS 分析显示,分别有28%、16%和 11%的病例存在 STAT5B、JAK2 和 JAK1 突变。Morsia 等发现,17 例CEL-NOS患者中80%存在突变,包括 ASXL1(42.9%)、IDH1(28.6%)以及其他基因,如 TP53、SRSF2、SH2B3、STAT5B、KDM6A 和 NF1。另一项研究描述了 12 例CEL-NOS患者的细胞遗传学和分子学特征,发现所有患者在初诊时均有 STAT5B 突变,其中 92%存在 STAT5BN642H突变;常见的共突变包括 SF3B1、SRSF2 和 TET2,STAT5BN642H也与CEL-NOS患者的嗜碱性粒细胞增多有关。
诊断标准
2022 年WHO和ICC发布了血液系统疾病的最新分类,对ED和CEL-NOS做出了关键改变(表 2)。这两种分类都需要克隆性和异常骨髓形态(如巨核细胞或红细胞发育不良)的证据来诊断CEL-NOS,同时排除MLN-TK中出现的BCR::ABL1和酪氨酸激酶基因重排。此外须排除其他伴有嗜酸性粒细胞增多的髓系肿瘤,如MPN、MDS、MDS/MPN、AML和系统性肥大细胞增多症(SM)。
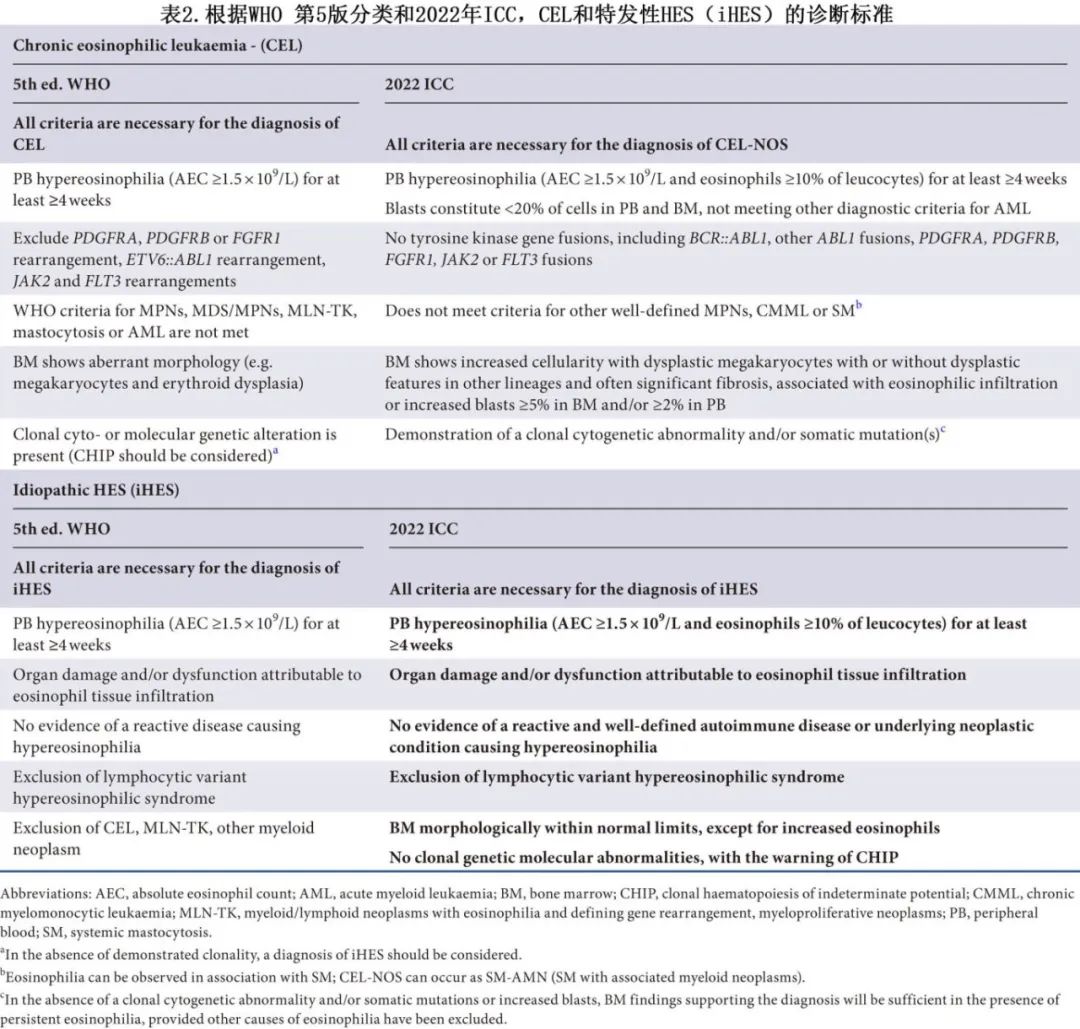
WHO和ICC的框架大致一致,但也存在显著差异。WHO已将持续性HE的持续时间从6个月缩短为4周。与ICC不同的是,WHO还取消了将骨髓或外周血原始细胞增多作为CEL-NOS的替代诊断标准的要求。ICC则维持这一标准,并要求外周血中嗜酸性粒细胞≥10%,且允许原始细胞增多(骨髓≥5%或外周血≥2%)作为支持性证据,前提是骨髓形态无异常。
鉴别诊断
反应性HE与器官损伤评估
HE的病因多种多样,包括反应性疾病和肿瘤性疾病。反应性形式是由可识别的因素触发的,这些因素会导致IL-5介导的嗜酸性粒细胞增殖(图 3)。寄生虫感染,尤其是由蛔虫、绦虫和吸虫引起的感染,是全球范围内继发性嗜酸性粒细胞增多最常见的病因。其他因素包括常见的过敏性/特应性疾病(例如哮喘、过敏性鼻炎和特应性皮炎)、药物(例如抗惊厥药、抗生素、磺胺类、抗风湿药和别嘌醇)、自身免疫性疾病(例如嗜酸性肉芽肿性多血管炎、类风湿性关节炎和慢性炎症性肠病)以及实体瘤的副肿瘤表现。
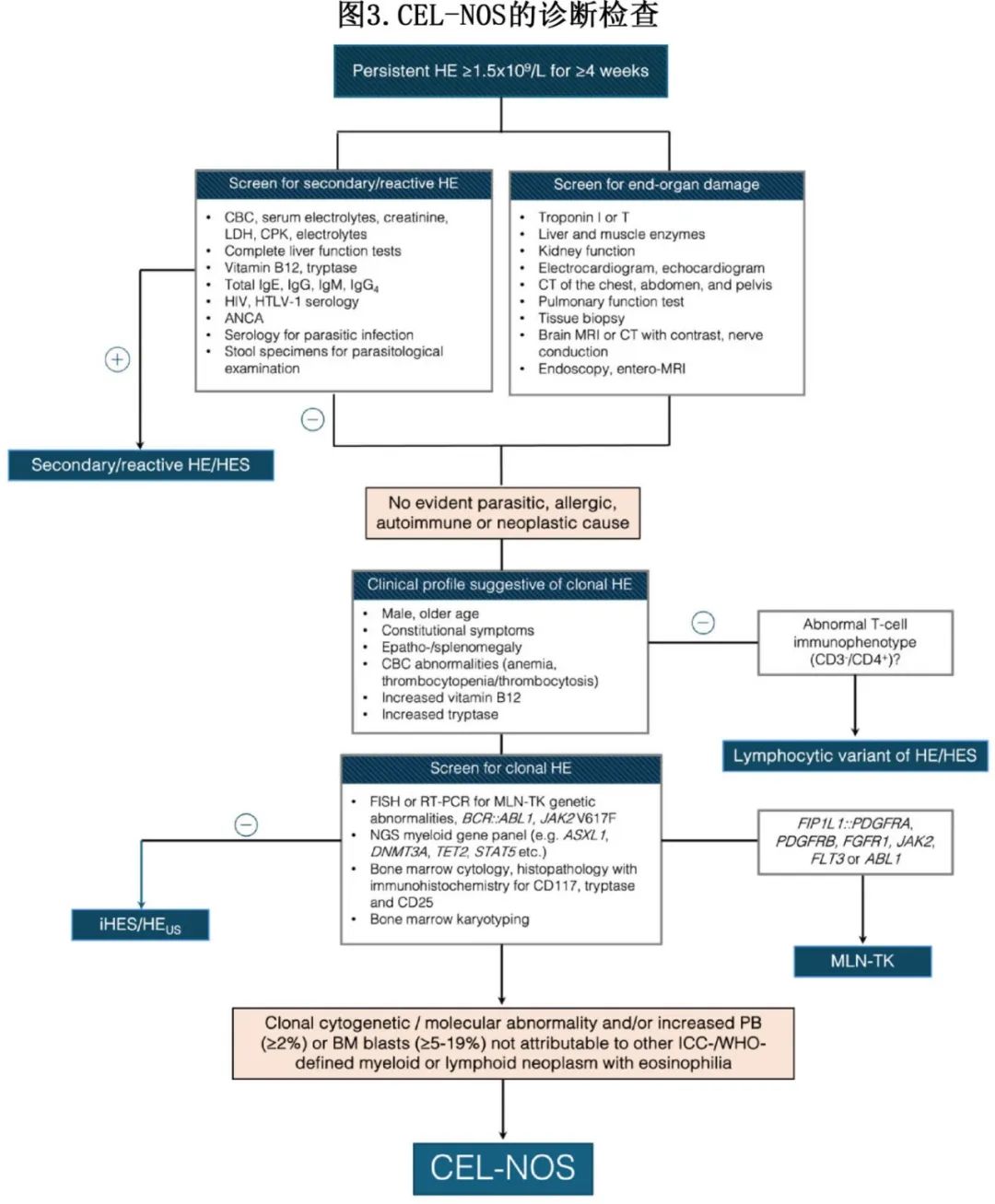
全面的患者病史对于诊断反应性嗜酸性粒细胞增多症的基础原因至关重要,包括近期旅行、药物使用和恶性肿瘤风险因素,以及血清学检测、粪便分析和自身免疫性血清学检测(表3)。如果嗜酸性粒细胞增多症在初步评估后仍持续存在,应根据临床判断进行进一步的诊断步骤,包括额外的血清学检测、影像学检查、内镜检查和组织学检查。
无论病因如何(家族性、继发性还是肿瘤性),全面的评估对于识别由于嗜酸性粒细胞组织浸润和细胞毒性蛋白沉积(HES的标志)导致的器官损伤或功能障碍都至关重要;该评估应包括全血细胞计数、外周血涂片、血清胰蛋白酶、维生素 B12 水平、免疫球蛋白 IgE 和心肌肌钙蛋白。此外,推荐的诊断成像和检查包括心电图(ECG)、超声心动图、肺功能测试以及胸部和腹部CT。
与嗜酸性粒细胞增多相关的血液系统肿瘤
一旦排除HE的继发性/反应性病因,诊断时应考虑 MLN-TK、CEL-NOS、与血液恶性肿瘤相关的克隆性嗜酸性粒细胞增多症、HE/HES (LV-HE/HES)和iHES/HEUS的淋巴细胞变体。
对克隆性嗜酸性粒细胞增多症的初步评估应包括对外周血和骨髓形态的评估。然而仅依赖嗜酸性粒细胞形态可能会产生误导,因为细胞学异常本身缺乏足够的特异性来区分肿瘤过程和反应性形式。尽管如此,这些评估对于准确原始细胞计数以及排除其他血液恶性肿瘤(此情况下嗜酸性粒细胞增生是细胞因子过量生成或涉及特定谱系的结果)仍至关重要。例如非霍奇金淋巴瘤和霍奇金淋巴瘤会引起显著的炎症反应,属于前者;后者则包括MPN、MDS和慢性粒-单核细胞白血病(CMML)。系统性肥大细胞增多症(SM)也可表现为克隆性嗜酸性粒细胞增多症。SM呈现出多面性临床表现,从惰性(ISM)到晚期形式(如侵袭性SM、肥大细胞白血病[MCL]以及伴有髓系肿瘤的SM(SM-AMN)。尽管罕见,SM-AMN也可能与CEL-NOS相关。在这种情况下,如果符合CEL-NOS的形态学标准,则最合适的诊断为SM-CEL;否则就应归类为伴有嗜酸性粒细胞增多症的SM。
MLN-TK
诊断工作需要通过常规细胞遗传学评估、荧光原位杂交(FISH)、分子学检测(通过外周血或骨髓样本PCR或NGS)来排除 MLN-TK(表 3)。MLN-TK 源自多能造血干细胞(HSC),这是由于超过 70 种已识别的酪氨酸激酶重排所致,其中至少有六种酪氨酸激酶反复受累,包括 PDGFRA、PDGFRB、FGFR1、JAK2、ABL1 和 FLT3。这些亚型在临床上是异质性的,可能表现出类似于CEL-NOS、MPN、MDS/MPN、B 细胞/ T 细胞ALL或AML的特征。此外,在 MLN-TK 各类别中,HE 的发生频率各不相同。由于 4q12 染色体间质缺失导致 PDGFRA 隐匿性重排,从而产生 FIP1L1::PDGFRA 融合,这是最常见的分子学异常,通过 FISH 或实时 PCR(RT-PCR)检测效果最佳。涉及 PDGFRB 的重排次之常见,其中 t(5;12)(q33;p13)/ETV6::PDGFRB 易位是最常见的融合伴侣,尽管已描述了超过 30 种不同的融合基因。临床上,具有PDGFRA和PDGFRB重排的MLN-TK都可能在外周血和骨髓中表现为CEL样表型。更罕见的是,MLN-TK中的重排基因是FGFR1,有14个易位伴侣,其中t(8;13)(p11;q12)/ZNF198::FGFR1占近50%的病例。此外,还报道了涉及 9p24 染色体上 JAK2 的易位,其中 t(8;9)(p22;p24.1)/PCM1::JAK2 易位最为常见。这些易位会导致慢性或急变性血液肿瘤,伴有不同程度的嗜酸性粒细胞增多。2022 年WHO和ICC分类都引入了新的MLN-TK诊断类别,包括具有 FLT3 和 ETV6::ABL1 重排的类别。
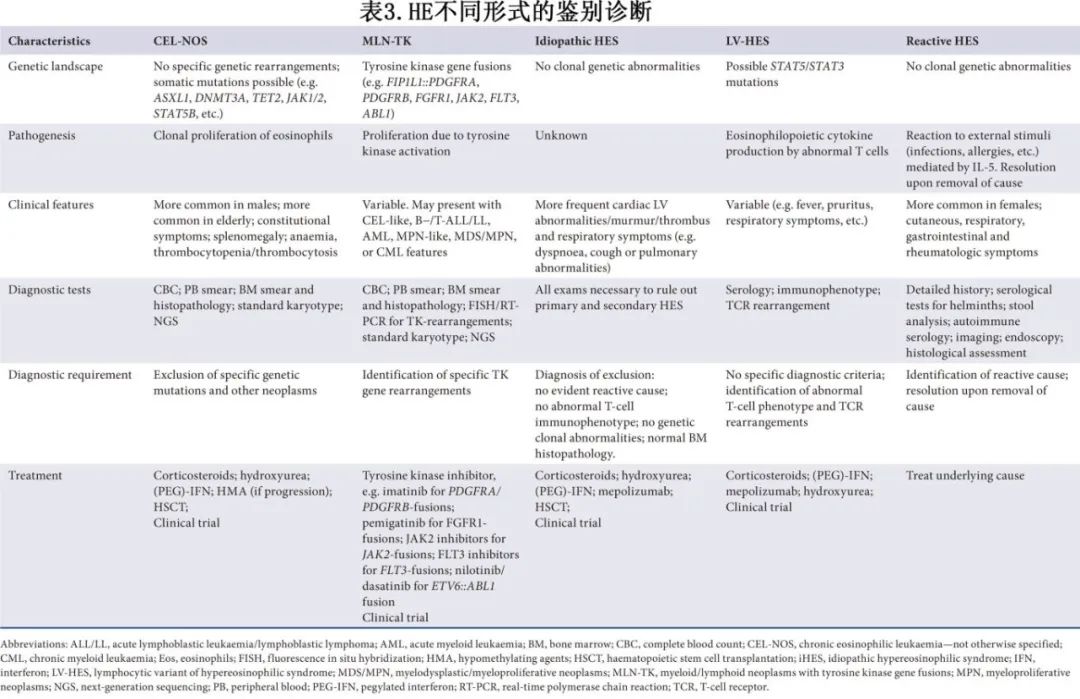
HE的淋巴细胞变体
淋巴细胞变异型嗜酸性粒细胞增多/嗜酸性粒细胞增多综合征(LV-HE/ HES)是一种生物学上独特且罕见的HE变体。在 LV-HE/HES 中,嗜酸性粒细胞增多是由于异常循环的 T 细胞产生嗜酸性细胞因子所致。这些细胞主要是克隆性的,通常具有异常的免疫表型,包括 CD2+/CD3−/CD4+/CD5+/CD7-/+/CD8−。其他表型也有报道,如 CD3+/CD4+/CD8−。尽管目前没有大规模的分子研究,但也报道了 STAT5 和 STAT3 突变。
临床上,LV-HES患者可表现为皮肤表现,包括皮炎、湿疹样病变、千皮病、荨麻疹斑块和血管性水肿,但其表现并不仅限于皮肤,还可包括呼吸系统、胃肠道和风湿系统症状。与CEL-NOS患者相比,LV-HE/HES患者中女性更常见,中位AEC更低,贫血和血小板减少症更少见,风湿系统症状更常见。鉴于与其他淋巴细胞增生性疾病有重叠特征,排除潜在的 T 细胞淋巴瘤或相关病症至关重要,如 IgG4 相关性疾病、皮肤和外周 T 细胞淋巴瘤(包括成人 T 细胞白血病/淋巴瘤和血管免疫母细胞性 T 细胞淋巴瘤[AITL])。诊断手段应包括 CT 或 PET 成像、受累淋巴结、骨髓或皮肤的活检以及外周血流式细胞术。尽管目前尚无明确的诊断标准,但确诊通常是基于存在具有异常免疫表型的T细胞群以及多存在T细胞受体(TCR)的单克隆重排。据报道,包括 AITL 在内的 LV- HES 中多达 25%会进展为 T 细胞淋巴瘤,因此准确的诊断评估不仅对于启动适当的治疗至关重要,对于将患者纳入监测项目以发现明显的恶性病变也同样关键。
特发性HES和意义未明的HE
从历史上看,由于在细胞遗传学正常的HE病例中难以证明克隆性,因此鉴别iHES和CEL-NOS一直具有挑战性。如前所述,NGS使得在原本被归类为特发性的病例中检测到突变成为可能。研究显示,与 NGS 检测到突变的病例相比,未检测到 NGS 突变的 iHES 患者更年轻,且更少出现贫血,而与嗜酸性粒细胞激活相关的症状则更常见,如皮肤病变、水肿、风湿症状或超敏反应。此外,骨髓细胞结构与年龄相适应,伴有罕见的巨核细胞异型性、红细胞生成(dyserythropoiesis)和粒细胞生成异常(dysgranulopoiesis)。在ICC分类中,将异常骨髓组织病理学纳入CEL-NOS的诊断标准(表2)增强了鉴别iHES的可靠性。
无器官损害的持续性HE患者(无法归因于反应性或肿瘤性原因)定义为HEUS。在这些病例中,分子检测也可以识别DNMT3A、TET2和ASXL1(DTA)等基因的突变;这些突变更表明为具有潜能未定的克隆性嗜酸性粒细胞增多症(CEIP),可能会增加患血液肿瘤的风险。这给iHES/HEUS患者的诊断和管理以及NGS中克隆性的证据带来了挑战。目前尚不清楚这些突变是否表明向更侵袭性形式的进展。这些患者相对较老的年龄表明,DTA突变可能反映与造血老化相关的克隆性扩增,使它们难以归类为CEL-NOS。值得注意的是,像SRSF2或SF3B1这样的剪接因子的突变可能比DTA突变扩增得更快,从而引发了更多担忧。鉴于这些突变对临床结局的影响不确定,建议对这些患者进行密切监测。
预后与风险分层
传统上认为CEL-NOS患者的预后极差,中位总生存期(OS)约为 1-2 年。然而,来自监测、流行病学和最终结果(SEER)数据库中 373 例CEL-NOS 患者以及国家癌症数据库(NCDB)中 588 例患者的分析数据表明,生存率比之前预期的要好。根据 SEER 数据,CEL-NOS 患者在 1 年、3 年和 5 年时的相对生存率分别为 88.8%、83.3%和 78.7%,而 NCDB 中 1 年、5 年和 10 年生存率分别为 86%、70%和 56%。另一项纳入487例 CEL-NOS 患者的 SEER 分析也得出类似结果,显示 5 年疾病特异性生存率为 95.6%,而 CEL-NOS 相关死亡的中位生存期仅为 5 个月。这些发现表明存在一个疾病更具侵袭性、预后明显更差的亚组,同时也有预后更良好的患者。鉴于文献的结果存在差异,需要进行进一步的分析,同时旨在开发有效的预后分层模型。由于其罕见性,缺乏用于指导特定治疗策略的标准化风险分层模型,然而最近的研究已经开始确定潜在的预后变量。通过分析 SEER 数据发现,年龄、诊断时的婚姻状况和继发性CEL与生存率显著相关,从而开发出列线图来预测 CEL-NOS 患者的 3 年和 5 年总生存率,有效地将其分为低危和高危组。有学者对 SEER 和 NCDB 的分析发现,年龄≥65 岁以及非学术中心诊断或治疗是生存率较差的预测因素,凸显了这种罕见且侵袭性疾病早期诊断和获得专门治疗中心治疗的关键性。此外,诸如巨核细胞非典型性和外周血嗜酸性粒细胞增多等形态学异常也成为生存率降低的预测因素。
由于对常规化疗反应不佳,CEL-NOS患者进展为AML是另一个重大担忧。文献报道的进展率各不相同:三个病例系列描述了相对较高的进展率,范围从 17%到 50%;而另一项研究中进展率较低,只有不到 1%的队列发生白血病转化。进展率的异质性可能是由于样本大小和患者选择的差异。目前关于进展为 AML 的潜在预测因素的数据非常有限。有研究的单因素分析显示,外周血嗜酸性粒细胞异常、显著纤维化、复杂核型以及对治疗无反应与白血病转化有关;然而由于样本量有限,这些因素在多因素分析中未确认为独立变量。
总的来说,整合临床和形态学参数可以提高CEL-NOS患者的生存风险分层和白血病进展风险分层。此外,突变检测可能有助于识别生存率较差的亚组,但还需要进一步研究来验证这些方法的预后潜力。
治疗
一般的治疗方法
HE的治疗存在临床挑战,需要多学科的诊断和治疗方法(图4)。
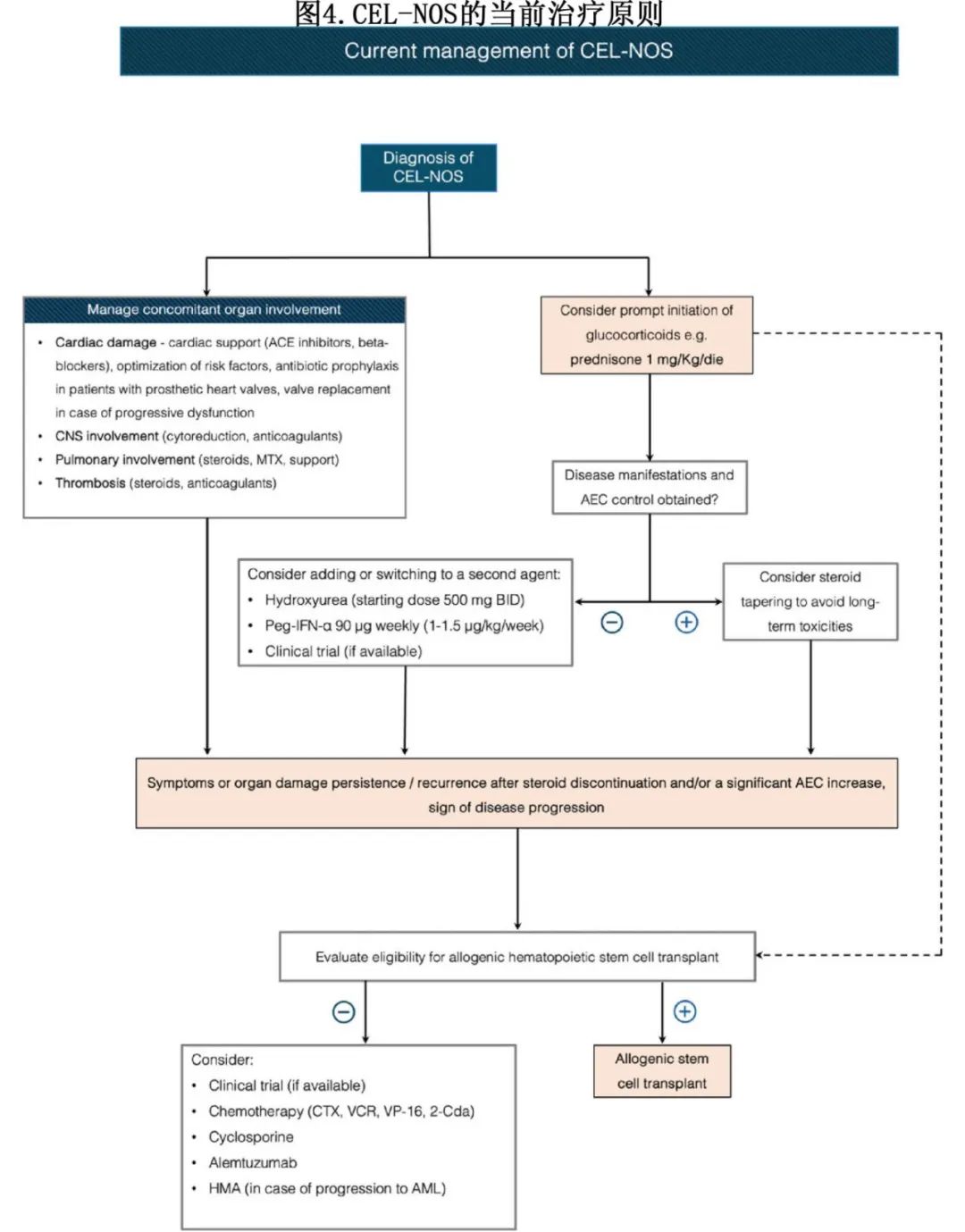
准确的诊断和识别潜在原因至关重要,尤其是在靶向治疗的时代。一旦排除反应性/继发性原因,则识别酪氨酸激酶重排必不可少,尤其是定义MLN-TK的重排,特别是PDGFRA和PDGFRB重排,因为它们对伊马替尼等小分子抑制剂敏感。相比之下,LV-HE的患者通常对类固醇耐药,对其他药物如干扰素α(IFN-α)和环孢素反应不一。
CEL-NOS患者缺乏标准化的治疗,且对于最佳一线治疗方案缺乏共识,从而进一步使临床管理变得复杂。一般来说,HE的治疗目标是降低嗜酸性粒细胞计数,以缓解嗜酸性粒细胞活化的临床表现,积极治疗旨在防止疾病进展和器官损伤,此外还希望实现骨髓原始细胞减少,以及细胞遗传学和/或分子学反应。
无论病因如何,HE都可能导致严重的、可能危及生命的并发症,包括心脏、呼吸系统、神经系统和血栓栓塞。是否开始治疗取决于临床表现的严重程度和急迫性,以及是否存在器官损伤。在出现危及生命的表现时不要延迟治疗。此外,由于无法排除寄生虫感染,使用伊维菌素进行经验性治疗(每天200μg/Kg,持续至少 2 周)也是合理的。
一些专家建议,鉴于器官损伤的可能性较高,应将AEC达到 1.5-2×109/L 视为开始治疗的阈值。然而在没有器官疾病的情况下,仅基于特定的嗜酸性粒细胞计数来启动治疗尚显证据不足。对于HEUS,定期监测的观察等待或可作为选择。而对于CEL-NOS患者,鉴于预后不良,即使没有器官功能障碍也应进行治疗,这一点已达成共识。
类固醇疗法
除了反应性/继发性疾病、伴 PDGFRA 或 PGDFRB 突变的 MLN-T K 以及重叠综合征外,皮质类固醇(CS)治疗长期以来一直是HES治疗的基石(图 4),包括在CEL-NOS中超过 80%广泛使用该治疗。CS 治疗对嗜酸性粒细胞的存活和功能可产生直接影响,并且对细胞因子网络的干扰可能间接有助于它们的发育、存活和功能。尽管有关于类固醇治疗 HE 疗效的若干报告,但治疗的剂量和持续时间仍不清楚。
考虑到第一个问题,对 188 例HES患者的回顾性分析中,糖皮质激素(CS)每日中位剂量为 40mg( 5-625mg)。此外,在接受类固醇治疗的 141 例患者中,85%实现完全或部分缓解。尽管关于CEL-NOS的具体数据有限,但专家们普遍建议考虑中高剂量CS,例如泼尼松每日1mg/kg。
关于治疗持续时间,>50%的CEL-NOS患者接受CS维持治疗。在某的研究中,治疗持续时间从 2 个月到 20 年不等。值得注意的是,对于需要长期治疗以控制嗜酸性粒细胞计数和症状管理的患者,CS 治疗可能会因副作用而变得复杂。因此一旦症状得到控制,且AEC降低到 1.5 倍正常水平以下,逐渐减少治疗似乎是合理的。
关于结局,对 NCDB 数据的分析表明,588例CEL-NOS患者中有 210 例接受CS治疗。尽管该研究未报告治疗方案或缓解率,但接受类固醇治疗的患者中1 年OS为 90%,5 年OS为 76%;而非类固醇治疗的患者中分别为 84%和 66%。
尽管CS疗效显著,但仍有 40%的患者因耐药而中断治疗。有人认为,嗜酸性粒细胞对类固醇治疗的耐药可能是由于糖皮质激素受体异常或IL-5水平升高导致细胞凋亡受损所致。在类固醇停药后症状复发、出现器官损害迹象和/或泼尼松剂量>10mg/天时嗜酸性粒细胞计数显著增加的情况下,可以考虑添加或换用其他药物。
羟基脲
羟基脲初始剂量为 500mg,每日两次,是治疗HES的有效药物,可单药或联合CS。在 188 例HES 患者中给予64例患者羟基脲降细胞治疗,其中 18 例接受单药治疗。中位每日剂量为 1000mg(500-2000mg)。在 18 例接受单药治疗的患者中,33%获得完全缓解(CR),39%获得部分缓解(PR)。47%的患者因耐药而停药,43%的患者因不耐受而停药。羟基脲联合CS时高达 69%的患者获得完全或部分缓解。因此,增加羟基脲进行降细胞治疗似乎是增加CS剂量的合理替代方案。
干扰素-α
聚乙二醇化干扰素-α(Peg-IFN-α)和非聚乙二醇化干扰素-α均可诱导克隆性HE患者血液学和细胞遗传学反应。
根据对文献的审查,42%的CEL-NOS患者使用了 IFN-α。初始剂量或维持剂量尚未明确界定,但建议每周 90μg(1-1.5μg/千克/周)聚乙二醇 IFN-α,或初始剂量每周三次皮下注射 100 万单位IFN-α并逐渐增加到每周三次 300-400万单位,以控制AEC。
联合CS治疗在缓解率方面显示出优势。在之前提到的一项多中心研究中,188例HES患者中有 46 例接受中位剂量为 1400 万单位/周的 IFN-α治疗;单药治疗的患者中有50%实现CR或PR,而接受联合治疗的患者中为75%。联合治疗在CS剂量降低方面也具有益处。在法国小组对 29 例 HES 患者进行的一项回顾性研究中,包括 4 例克隆性HE患者,16 例患者接受平均剂量为 113μg/周的聚乙二醇化 IFN-α治疗,13例患者接受平均剂量为 1200 万单位/周的非聚乙二醇化 IFN-α治疗。治疗 1 个月后,16例患者实现血液学完全缓解(CHR)。此外,IFN-α治疗与泼尼松的平均每日剂量显著减少相关。
然而IFN-α 治疗也有副作用。在已报道的研究中停药率很高。就生存率而言,纳入 NCDB 研究的 17 例(3%)患者接受IFN-α 治疗,但免疫治疗与总生存期改善无关。
甲磺酸伊马替尼
甲磺酸伊马替尼目前是伴有 PDGFRA 和 PDGFRB 重排的MLN-TK的一线治疗药物,但部分HES和CEL-NOS患者可能对伊马替尼有反应,通常剂量高于每日 400mg。事实上,过去有报道称,58.3%的CEL-NOS患者使用了伊马替尼。一项研究在PDGFRA 阴性HES中评估伊马替尼反应的预测因素,包括外周血嗜酸性粒细胞增多伴有发育不良、贫血和/或血小板减少症、骨髓细胞过多、骨髓活检中发育不良的肥大细胞或巨核细胞、血清类胰蛋白酶水平≥12ng/mL以及网状纤维≥2 等因素。具有 KITM541L 突变的CEL-NOS对每日 100mg伊马替尼敏感,可实现持续的完全缓解,中位随访 74 个月。尽管如此,不同研究中的反应仍然异质性较大,完全缓解病例可能表明存在伊马替尼敏感靶点,但尚未被确定。
化疗与异基因移植
关于CEL-NOS患者的化疗缺乏全面报道,大多数信息来自病例报告和一小部分HES患者的系列研究。已有关于长春新碱、环磷酰胺、依托泊苷、2-氯脱氧腺苷联合阿糖腺苷、环孢素的反应的报道,也有报道在CEL-NOS进展为AML的情况下使用去甲基化药物。然而由于缺乏针对CEL-NOS的大规模研究和具体数据,无法对该患者群体的化疗疗效进行精确和明确的评估。
英国血液学标准委员会(British Committee for Standards in Haematology)关于ED的指南强调了造血干细胞移植(HSCT)的作用有限。根据其适应症,HSCT 适用于伴有 FGFR1 重排的MLN-TK患者、CEL-NOS患者,以及对常规TKI不耐受或难治性或发生进展性器官损害的患者。2022 年,欧洲血液和骨髓移植(EBMT)小组公布了一项回顾性研究的结果,这是目前关于 CEL-NOS 患者移植的最大经验。该研究纳入77 例HES(n=47)或 CEL-NOS(n=30)的成年患者接受异基因造血干细胞移植,采用清髓性预处理方案(MAC),主要包括白消安和环磷酰胺,52%的HES患者和61%的CEL-NOS患者使用该方案。最常见的减强度预处理(RIC)包括氟达拉滨和苯达莫司汀。67%的CEL-NOS患者和79%的HES患者的主要干细胞来源为外周血。非复发死亡的最常见原因为移植物抗宿主病(GVHD)和感染性并发症。总体而言,CEL-NOS患者的生存率显著低于HES患者,1年和3年总生存率分别为46%和34%,而HES患者分别为69%和59%。尽管移植是唯一可能治愈的选择,但CEL-NOS患者往往年龄较大,合并症较多,从而限制了其接受移植的机会。因此,确定CEL-NOS患者的理想移植候选者和时机仍然具有挑战性,尚未明确界定。
Alemtuzumab
嗜酸性粒细胞表面 CD52 表达的证据促使对抗 CD52 单克隆抗体Alemtuzumab在HES和CEL-NOS中进行评估。基于难治性 HES 的良好缓解,进一步对 12 例特发性 HES 和难治性 CEL-NOS 患者进行测试,剂量为 5-30mg每周 1-3 次。尽管 12 例患者中有 10例实现CHR,但 11 例患者复发。其中6例患者再次接受Alemtuzumab治疗,5 例实现第二次 CHR。尽管这些数据有限,但副作用以及为维持反应而进行长期治疗的需求限制了其在临床实践中的应用。
其他药物和新视角
鉴于IL-5在嗜酸性粒细胞的分化、存活和激活中起着关键作用,针对该细胞因子的单克隆抗体在HES的治疗管理中也引起了极大关注。目前FDA和EMA批准的唯一用于治疗 HES 的 IL-5 抑制剂为mepolizumab,其抑制IL-5 与嗜酸性粒细胞上表达的 IL - 5 受体α链之间的相互作用。临床研究表明其有诸多获益,包括减少嗜酸性粒细胞计数(AEC)、降低CS剂量,以及缓解瘙痒、皮肤病变、鼻息肉和吞咽困难等症状。尽管这些成果显著,但在克隆性HES(包括CEL-NOS)的使用方面仍存在重大差距。FIP1L1::PDGFRA 阳性 HES 患者通常排除在主要临床试验之外。由于缺乏数据,mepolizumab抗和其他 IL-5 抑制剂尚未纳入 CEL-NOS 的治疗方案中。
尽管治疗选择有限,但文献显示一些临床前研究探索了新的CEL-NOS治疗药物。一份病例报告展示了一例17 岁 CEL-NOS 患者的数据,其中 NGS 分析鉴定出 JAK1 R629_S632delinsSA 突变,其变异等位基因频率(VAF)为 23%,另外还有两个胚系突变,分别为 SETD2 R136T(VAF 50%)和 T2421A(VAF 47%)。Ba/F3 转化细胞表现出高水平的 STAT3 和 STAT5 磷酸化,但在芦可替尼治疗后显著减少。然而血液和骨髓样本对托法替尼和 JAK1 抑制剂显示出部分敏感性,但对芦可替尼耐药,表明需要进一步研究靶向治疗。目前正在进行一项 II 期研究,以评估芦可替尼在HES或原发性ED患者中的疗效,包括 CEL-NOS(NCT03801434)。
2021 年开展了一项临床前研究,利用表达抗嗜酸性粒细胞单克隆抗体(AAVrh.10mAnti-Eos)的腺相关病毒(AAV)开发基因疗法,旨在实现持续的嗜酸性粒细胞支持。通过给药表达IL-5细胞因子的AAV,建立新的NOD-scid IL2rγnull小鼠模型,导致各器官和组织中明显的外周和组织嗜酸性粒细胞增多,导致死亡。与对照组相比,给小鼠模型注射AAVrh.10mAnti-Eos可显著降低嗜酸性粒细胞水平和死亡率。
结论与未来方向
CEL-NOS由于治疗选择有限、预后不良以及进展为急性白血病的风险,带来了诊断和治疗上的挑战。细胞遗传学和分子生物学的进步完善了HE和HES的诊断标准,从而确定了可采用靶向治疗的CEL亚型(MLN-TK)。然而CEL-NOS仍缺乏可识别的遗传驱动因素,没有特定的干预点。患者通常是合并多种疾病的老年人,移植治疗的机会有限,替代疗法只能带来短期反应,且副作用显著。进一步研究髓系突变(如 ASXL、TET2、DNMT3A)的预后意义,并开发新的靶向治疗以增加治疗选择至关重要。为 CEL-NOS 制定具体指南以及促进医疗保健提供者持续教育极为紧迫,以优化诊断、治疗和预后。
参考文献
Costa A, Scalzulli E, Breccia M. Chronic eosinophilic leukaemia—Not otherwise specified: Clinical features, genomic insight and therapeutic strategies. Br J Haematol. 2024;00:1–17.https://doi.org/10.1111/bjh.19921

