Nat Commun:基于UKB和All of Us队列的串联重复序列变异分析,揭秘人类基因组“遗传性缺失”
时间:2024-12-21 11:01:42 热度:37.1℃ 作者:网络
导读
在过去二十年中,已有数千项全基因组关联研究(GWAS)识别了影响各种人类特征的遗传变异,但多数研究普遍关注于二等位基因单核苷酸变异(SNV),对于可能包含多等位基因变异且突变率反复或高突变率的复杂基因组位点,GWAS的检测结果通常较差,这被认为是导致所谓“遗传性缺失”的原因。
作为一种未被充分研究的遗传变异类型,串联重复序列(TRs)可能有助于解释部分“遗传性缺失”问题,其包括短串联重复序列(STRs)和长串联重复序列(LTRs)两类。STRs的极端扩增已被认为是罕见神经退行性疾病和先天性疾病的原因;近期研究也表明,STRs和具有较大基序TRs的共同长度多态性可对基因组产生功能影响,导致基因表达、剪接和DNA甲基化的个体间差异。但由于缺乏能够进行大规模基因分型的技术,TRs经常被忽视。
近日,美国西奈山伊坎医学院的科研团队在Nature Communications发表了题为“A phenome-wide association study of tandem repeat variation in 168,554 individuals from the UK Biobank”的文章,以探究TRs多等位基因变异对人类性状的影响。研究团队对英国生物样本库(UKB)168,554名个体进行TRs基因分型分析和全表型组关联研究(PheWAS),共识别出47个与73个性状表现出精细映射关联的TRs;并在All of Us队列中验证了31个关联中的23个(74%),发现新关联可归因于TR长度的常见多态性变异,而非罕见的扩增。此外,精细映射的TRs也与局部基因表达和DNA甲基化密切相关。综上,该研究强调了多等位基因TR对人类基因组“遗传性缺失”的潜在贡献。

文章发表在Nature Communications
1.UKB中TRs的全表型组关联研究
研究团队首先鉴定了48,913个TRs,97%的基序大小在2-6bp之间,8%与基因编码区或UTR重叠,46%与带注释的调控元件重叠;并对UKB队列中168,554名无亲缘关系的欧洲血统个体进行基因型分析,得到了上述TRs的基因型。此外,研究团队还对All of Us队列1,027个个体中的同一组TRs进行了基因分型,以确保只保留具有高质量基因型的TRs。经筛选,最终保留36,085个高质量多态性TRs,用于对UKB队列中30,291个二元、定量和分类性状进行PheWAS。
通过将每个TR的平均等位基因大小与每个性状联系起来,共观察到5,378对显著TR:性状关联。接下来,研究团队使用两种互补的方法,即使用CAVIAR进行统计精细映射以及对TR和SNV基因型同时进行条件分析,来识别TR是最有可能导致观察到的性状关联变异的位点子集;最终定义了101个TR:性状对,具有高置信度的精细映射关联。
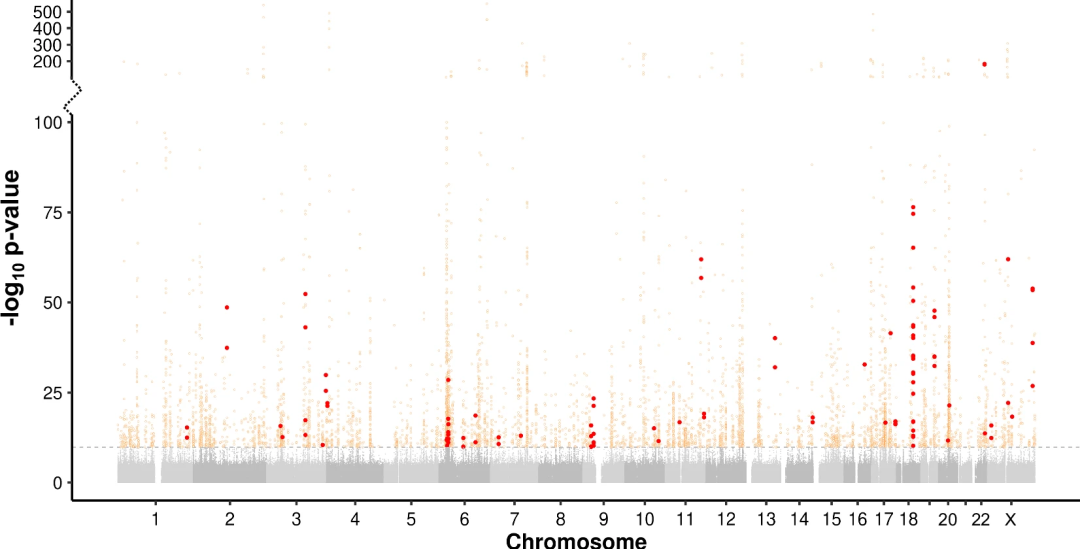
图1.曼哈顿图显示出36,085个TR和30,291个性状之间的PheWAS结果
2.新的精细映射关联由TRs的常见多态性变异驱动,而非极端扩增
上述高置信度精细映射关联集包括8个已知能够罕见扩增到异常大的TRs,其中6个已被证明会导致显性重复扩增障碍(REDs),另外两个(BCL2L11和 CBL)致病性不确定。研究团队对其中4个REDs进行分析,发现通过PheWAS鉴定出的相关性状与已知的极端扩增的表型效应一致,这表明该研究正确鉴定出了与TR长度的真正因果关系:包括亨廷顿病(HD)与HTT中编码的CAG重复序列长度增加相关,运动神经元疾病导致的死亡与C9orf72中内含子的GGCCCC重复相关等。
为验证其他精细映射关联是否同样可归因于罕见的扩增TR等位基因,研究团队进行重复关联检验。结果显示,除4个REDs外,几乎所有其他精细映射关联都保留了显著性,表明其均归因于常见的等位基因变异,而非罕见的扩增等位基因,包括TCF4重复序列长度与手臂阻抗、白细胞计数和淋巴细胞计数之间的新关联。
特别地,在新精细映射关联中还有三个TRs会发生罕见扩增,导致等位基因甲基化、转录沉默伴有叶酸敏感的脆性位点,即AFF2的5'UTR的poly(GCC)基序、BCL2L11的5'UTR的poly(CGC)基序和CBL的5'UTR的poly(CGG)基序。与TCF4一致,这些TRs的长度变异与血细胞性状之间的关联并不是极端扩增的结果,而由常见的多态性变异所致。
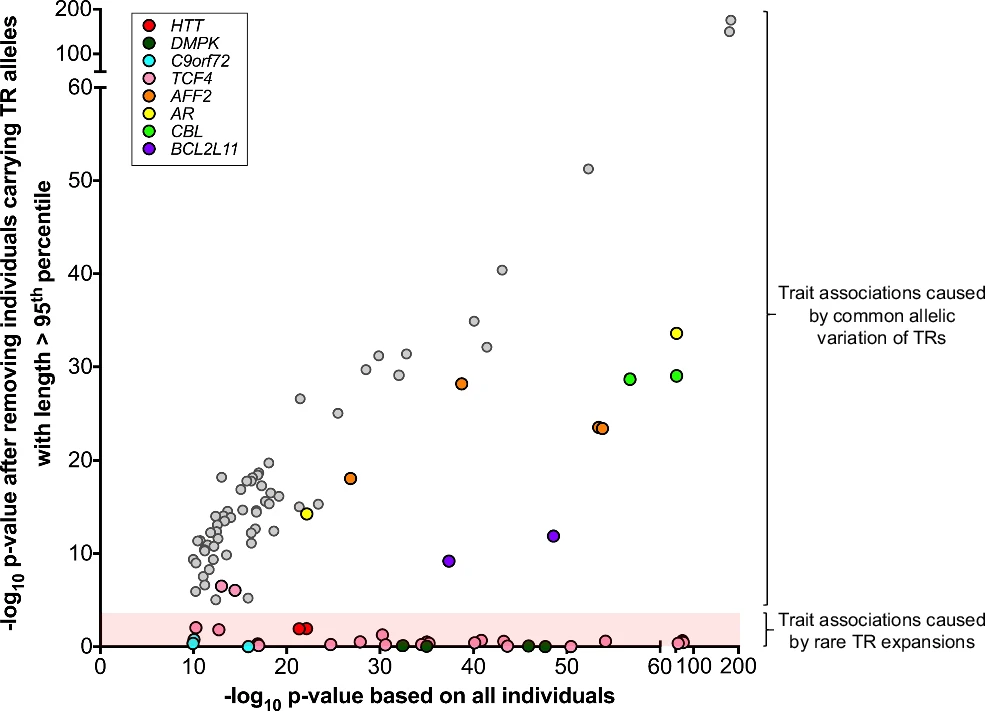
图2. 新的TR关联由常见等位基因变异而非罕见扩增驱动
在新的精细映射关联中,值得注意的是HRCT1基因外显子1中编码poly(CCA)基序。该TR具有极高的多态性,可编码一个富含组氨酸的氨基酸序列,与高血压发病率和使用降压药呈强烈的负相关性。该TR带来的风险是,携带HRCT1 TR等位基因最短5%的个体患高血压的风险比携带最长5%等位基因的个体平均高11%。
此外,研究团队还观察到一些新精细映射TRs,其与先前通过PheWAS发现的具有相同性状的基因重叠,为这些TRs的功能作用提供了间接证据。例如,GNB2的5'UTR中一个poly(CGC)基序,与脉搏率相关;WNT9A的3'UTR中一个poly(AC)基序,与身高相关。
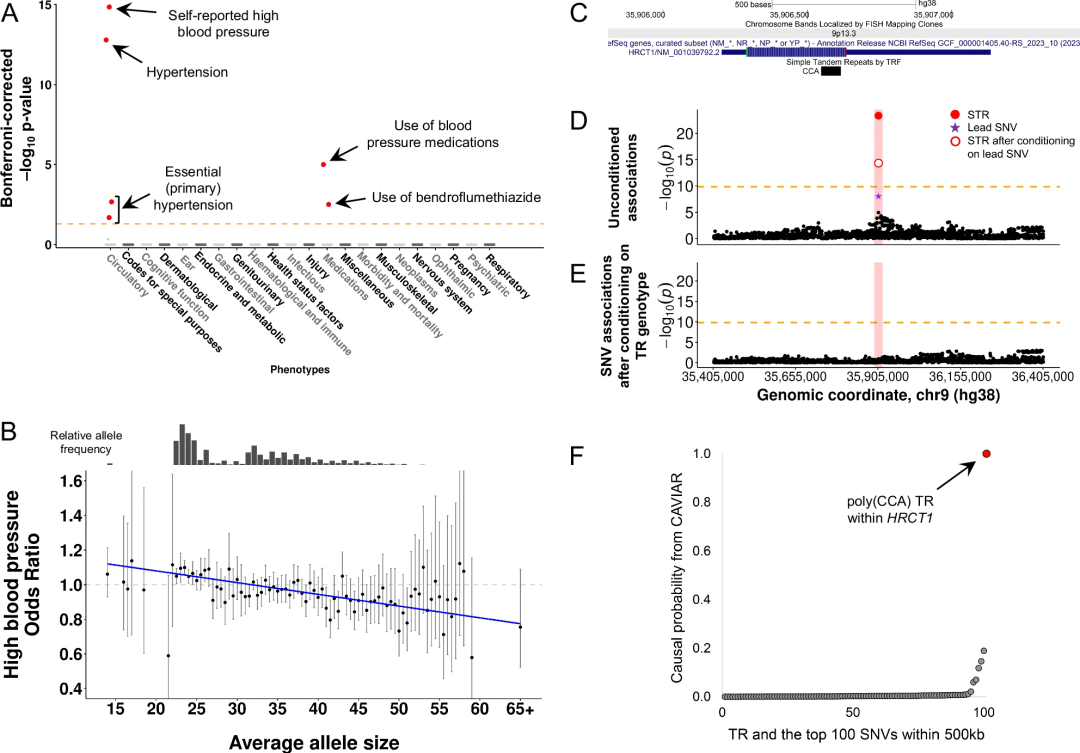
图3. HRCT1中高度多态性的编码CCA重复序列与高血压风险相关
3.All of Us队列的验证分析
研究团队通过All of Us队列验证了在PheWAS中确定的一组高置信度精细映射关联。首先确定了All of Us参与者的表型(与UKB相匹配),并具有足够的样本量对73个精细映射相关性状中的 31个进行复制分析;最终使用了88,406名All of Us个体GS数据中的TR基因型。在31个精细映射关联中,有23个具有显著性,并与在UKB队列中观察到的效应方向相同。
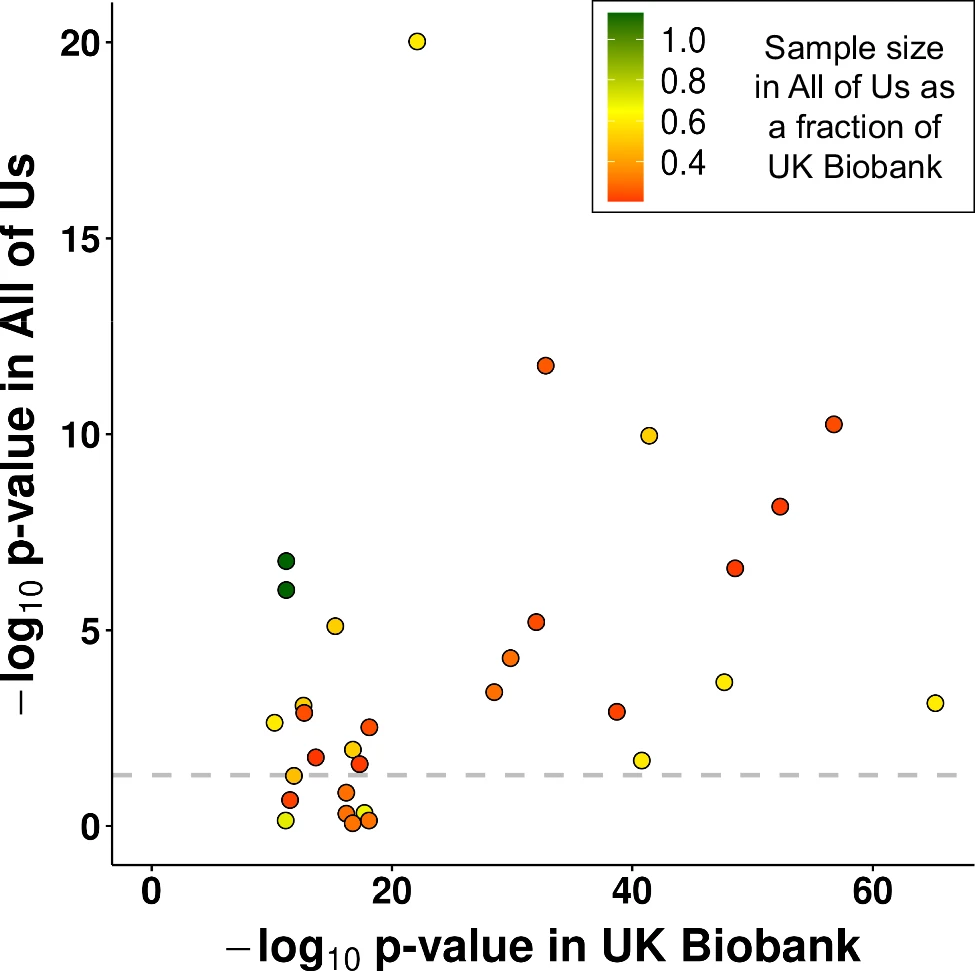
图4. All of Us队列中的验证分析
4.精细映射的TRs在基因表达和表观遗传学上具有较强功能效应
为评估TR变异对局部基因表达和表观遗传学的功能影响,研究团队从现有GS数据中对用于PheWAS的TRs进行基因分型,并将其与GTEx项目生成的49个组织的DNA甲基化和RNAseq数据进行比较,确定了作为表达或甲基化QTLs(eQTLs和mQTLs)的TRs。
结果显示,在UKB中通过PheWAS显示出显著性状关联的所有TRs,其在eQTLs中富集了4.9倍(相对于null(无效应)),在mQTLs中富集了3.5倍;被认为是性状关联高置信度精细映射变异的TR子集,其在eQTLs中富集了11.2倍,在mQTLs中富集了4.7倍。
例如,与高血压风险相关的HRCT1中的编码poly(CCA)重复序列,既与HRCT1表达呈负相关,又与HRCT1 3'UTR中CpG位点的甲基化水平呈正相关。其他多个精细映射TR也与重叠基因和/或顺式中其他基因的表达水平相关,表明该研究发现的许多TR关联的作用机制是通过改变基因表达和/或表观遗传学来实现的。
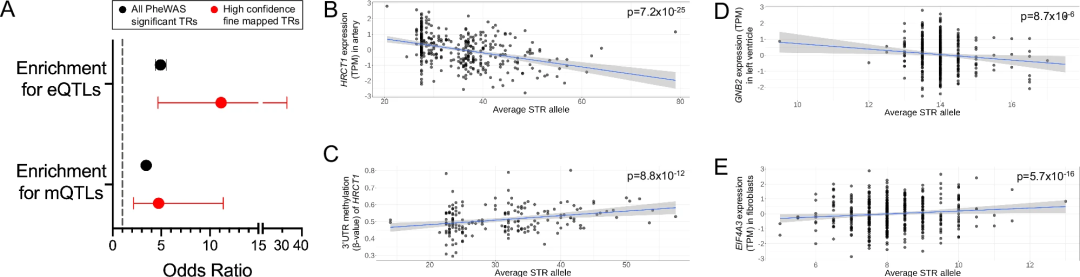
图5. 精细映射的TRs长度变化与局部基因表达和DNA甲基化相关
综上所述,研究团队利用现有基因组测序数据,首次对基因组中最具多态性和不稳定的TRs进行直接基因型分析和PheWAS,发现了许多与这些TRs长度变异相关的新表型关联;确定了与几种复发性RED相关的已知因果效应等。该研究表明多等位基因TR有助于解释基于SNV的GWAS“遗传性缺失”,强调了在精细映射研究中TR的重要性。
论文原文:
Manigbas, C.A., Jadhav, B., Garg, P. et al. A phenome-wide association study of tandem repeat variation in 168,554 individuals from the UK Biobank. Nat Commun 15, 10521 (2024).
https://doi.org/10.1038/s41467-024-54678-0

