CMI:电子科技大学王晨辉等团队研究明确复发性外周T细胞淋巴瘤的潜在治疗靶点
时间:2024-12-20 14:00:23 热度:37.1℃ 作者:网络
T细胞正常的细胞代谢对于有效的免疫反应至关重要。然而,当内源性或外源性代谢因子失调时,T细胞可能会导致癌症和自身免疫性疾病等多种疾病,仍不完全清楚T细胞的代谢调节机制。
2024年12月13日,电子科技大学王晨辉、肖雪、张存金、Du Yanyun共同通讯在 Cellular & Molecular Immunology在线发表题为“MYO1F regulates T-cell activation and glycolytic metabolism by promoting the acetylation of GAPDH”的研究论文,该研究表明Myo1f是TCR刺激后人类和小鼠T细胞激活所必需的,T细胞特异性MYO1F敲除小鼠的体内T细胞激活受损,肿瘤负荷增加且EAE严重程度减弱。
从机制上讲,TCR刺激后,MYO1F在酪氨酸607和634处被LCK磷酸化,这对于乙酰转移酶α-TAT1介导的Lys84、86和227处的甘油醛-3-磷酸脱氢酶(GAPDH)乙酰化是至关重要的,对于其活化、细胞糖酵解以及T细胞的效应子功能同样重要。重要的是,作者发现VAV1-MYO1F的融合蛋白(一种复发性外周T细胞淋巴瘤(PTCL)相关的致癌蛋白)促进GAPDH的高乙酰化及其活化,导致异常的糖酵解和T细胞增殖,抑制GAPDH活性显著限制了T细胞的活化和增殖,并延长了VAV1-MYO1F敲入小鼠的存活时间。此外,GAPDH的超乙酰化在含有VAV1-MYO1F基因融合体的人类PTCL患者样本中得到证实。总得来说,该研究不仅揭示了MYO1F调节T细胞代谢和VAV1-MYO1F融合诱导PTCL的机制,还表明PTCL是潜在的治疗靶点。

在感染和癌症发生过程中,初始T细胞在遇到同源抗原时被激活并成为效应T细胞,这个过程伴随着营养摄入和细胞代谢的剧烈变化。当再次遇到相同的抗原时,记忆T细胞会迅速“回忆”自身活性,同样伴随强烈的细胞代谢变化。适当的代谢过程改变可确保T细胞在不同的免疫反应中发挥适当的功能,从而增加T细胞对病原体和癌细胞的杀伤能力。已知几种信号分子参与TCR刺激诱导的T细胞代谢变化,如PI3K-AKT-mTOR轴、Myc信号传导和AMPK信号传导是TCR刺激后效应CD4+ T细胞和CTL产生和代谢重编程的主要调节因子。初始CD4+ T细胞分化为TH1和TH17细胞,依赖缺氧诱导因子1α(HIF1α)进行糖酵解,而TH2分化依赖于小GTP酶RHOA。
许多疾病中均能观察到T细胞代谢改变,相关学者认为其与许多疾病的发病机制有关,如癌症和自身免疫性疾病。乳酸直接抑制效应T细胞功能,乳酸积累抑制T细胞中的糖酵解和一碳通路,有利于肿瘤生长和免疫逃逸。GLUT1是T细胞中一种重要的糖酵解酶,GLUT1缺乏可减轻小鼠结肠炎和实验性自身免疫性脑脊髓炎(EAE,一种多发性硬化症小鼠模型)的严重程度,并用2-脱氧葡萄糖(2-DG)阻断糖酵解可改善EAE的预后。因此,靶向T细胞代谢在治疗许多疾病(如癌症和自身免疫性疾病)方面潜力巨大。
GAPDH是有氧糖酵解过程中的重要酶,在Warburg效应过程中起限速作用。除在糖酵解过程中发挥作用外,它还具有非糖酵解功能。GAPDH在不同的癌症类型中过表达,但其在癌症中的作用尚不完全清楚。GAPDH在T细胞活化和功能中起关键作用。如再遇抗原时,GAPDH的乙酰化迅速增加记忆CD8+ T细胞的活化和增殖,但参与这一过程的GAPDH乙酰转移酶是未知的。
GAPDH的过表达通过降低缺氧调节下HIF1α的表达来抑制T细胞增殖和存活。有研究表明,效应记忆(EM)CD8+ T细胞在早期表现出明显更强的GAPDH活性。一项研究表明,GAPDH与IFNG mRNA3′-非翻译区(UTR)中富含AU的元件结合,参与糖酵解过程并影响随后的IFNγ产生。有趣的是,小鼠T细胞中GAPDH的条件性过表达会导致外周Tfh样淋巴瘤,该淋巴瘤表型与人血管免疫母细胞性T细胞淋巴瘤(AITL)类似,是人PTCL的一种亚型。综上所述,既往研究表明GAPDH在调节T细胞功能中起关键作用,但仍不清楚GAPDH在T细胞中的调节机制。
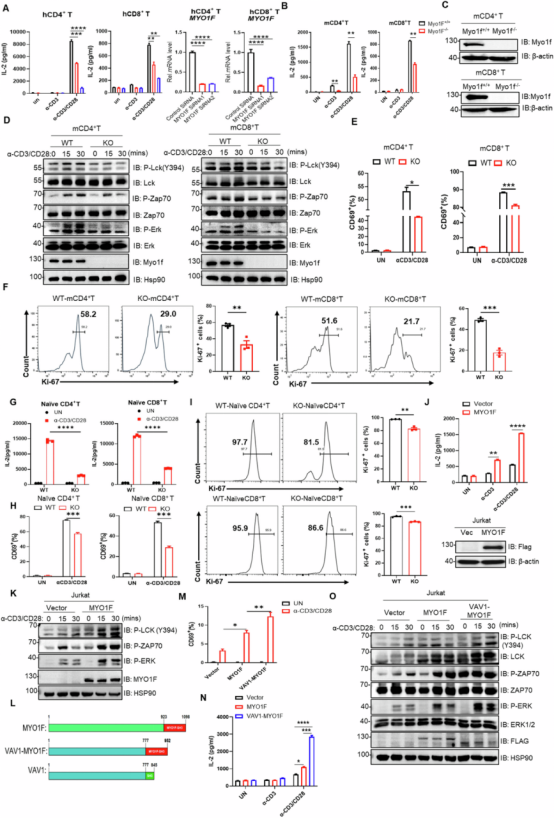
图1 MYO1F在体外调节T细胞活化(图源自Cellular & Molecular Immunology )
MYO1F是一种非常规肌球蛋白,仅在哺乳动物免疫细胞中表达。研究表明,MYO1F在宿主防御单核细胞增生李斯特菌和白色念珠菌中起关键作用,并调节免疫细胞的吞噬作用和生物行为。MYO1F在TCR刺激的人T细胞中高表达,MYO1F在各种人癌症样本浸润的CD8+ T细胞中高表达。有趣的是,经常在人外周T细胞淋巴瘤(PTCL)中发现MYO1F-VAV1基因融合基因,人MYO1F-VAV1在小鼠T细胞中的过表达会诱导恶性T细胞疾病,这种疾病会复制人PTCL表型。近期研究发现,MYO1F::WNK4在der(19)t(17;19)和VPS25::MYO1 Fonder导致骨髓增生异常综合征(MDS)。然而,尚不清楚MYO1F-VAV1和其他MYO1F融合蛋白影响PTCLs和MDS肿瘤发生的机制。
该研究表明,MYO1F是TCR刺激诱导的T细胞活化和增殖所必需的。从机制上讲,TCR刺激后MYO1F在酪氨酸607和634位点被磷酸化,对于其募集α-TAT1以在Lys84、Lys86和Lys227位点乙酰化GAPDH至关重要,促进T细胞活化和代谢变化。最后,作者发现MYO1F-VAV1致癌融合蛋白促进GAPDH的高乙酰化,这是导致异常T细胞活化和代谢变化的原因。抑制hMYO1F-VAV1敲入小鼠的GAPDH活性降低了T细胞活化并显著延长KI小鼠的存活。总体而言,该研究不仅确定了一种调节T细胞代谢的新机制,还发现了MYO1F-VAV1基因融合诱导PTCLs的机制,为PTCLs治疗提供潜在靶点。
参考消息:
https://www.nature.com/articles/s41423-024-01247-6

